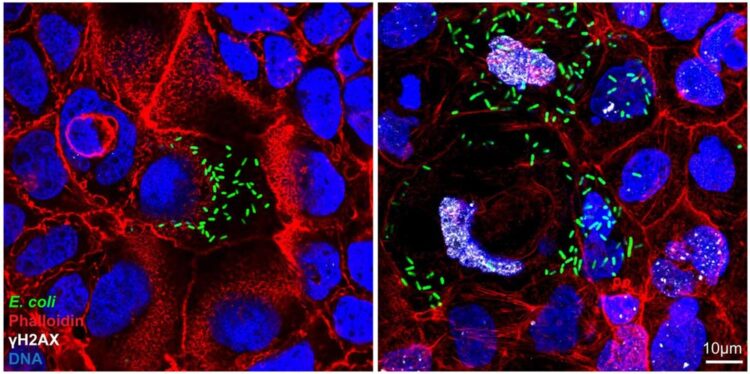
Escherichia coli bacteria are constitutive members of the human gut microbiota. However, some strains produce a genotoxin called colibactin, which is implicated in the development of colorectal cancer. While it has been shown that colibactin leaves very specific changes in the DNA of host cells that can be detected in colorectal cancer cells, such cancers take many years to develop, leaving the actual process by which a normal cell becomes cancerous obscure.
The group of Thomas F. Meyer at the Max Planck Institute for Infection Biology in Berlin together with their collaborators have now been able to “catch colibactin in the act” of inducing genetic changes that are characteristic of colorectal cancer cells and cause a transformed phenotype – after only a few hours of infection.
More than two-thirds of colorectal cancer patients carry colibactin-producing E. coli strains in their gut and the number of carriers is rising in the western world. Epidemiological evidence for a link between certain bacterial species and some forms of human cancer abound – but it remains difficult to provide the direct proof required to justify extensive prevention strategies. Meyer’s team recently provided the first definitive evidence for such a link by identifying the genetic signature colibactin leaves in host cells, and showing that it can be detected in a subgroup of colorectal cancers.
Now they have gone a significant step further by utilizing organoids to observe transformation itself. This new technology makes it possible to grow normal, primary colon epithelial cells in culture in the form of 3D spheres. These hollow “mini-organs” are generated by the adult stem cells that drive the rapid turnover of the colonic mucosa.
Prior to the advent of this technology, infection experiments in vitro required cell lines, which are already partially transformed and thus unsuitable for recapitulating the very early stages of cancer development. To test whether colibactin-producing E. coli have any lasting effect on host cells, the team infected their organoids for three hours. This was already sufficient to induce changes that are characteristic of colorectal cancer. Not only did the infected cells begin to proliferate faster than normal, but a subset of cells no longer required the presence of Wnt protein in the growth medium.
Growth factor drives stem cell turn-over
This critical “growth factor” is present in the environment surrounding the stem cells in the bottom of colon glands and drives their turn-over. Under healthy conditions, uncontrolled proliferation of the cells is prevented as soon as they leave this Wnt-containing niche. “Then they cease proliferation and take over digestive functions, only to be sloughed off once they reach the surface, pushed along by the continuous stream of cells leaving the stem cell niche,” says Michael Sigal, one of the senior authors who recently established his own laboratory at the Charité University Hospital in Berlin to study the phenomenon in greater detail. He further explains: “The same phenomenon can be observed in the organoid cultures: they require the continuous presence of Wnt to keep growing. Without it, the cells differentiate and die shortly afterwards.”
Such growth factor independence, as observed for the infected organoids, is a characteristic of early colorectal cancer cells. Sequencing of these organoids revealed that they contained numerous mutations, including large structural changes that led to whole sections of chromosomes being lost, gained or rearranged. “Surprisingly, we did not observe mutations in genes directly involved in Wnt signaling, which are known to cause colorectal cancer in patients who inherited such mutations. Instead, we found mutations related to p53 signaling”, says Amina Iftekhar, first author on the new paper. This important tumour suppressor is known as the “guardian of the genome” and so far, only a few studies had hinted at the possibility that it may also affect Wnt dependence.
Mutations in the p53 signaling pathway
Thomas F. Meyer explains that these findings fit well with the evidence from large cancer sequencing programs: “It is clear that colorectal cancer can arise through different mechanisms. In cases driven by chronic inflammation, such as Colitis or Crohn’s disease, where colibactin-producing E. coli strains are particularly prominent, mutations in p53 are indeed found to be an early event.” And the large chromosomal rearrangements they observed are found in the majority of colorectal cancer cases.
According to Meyer, this has important implications: “Although the majority of colorectal cancer patients carry colibactin-producing E. coli, we were puzzled by the fact that the colibactin signature can only be detected in a small proportion – up to ten percent. Our new results now suggest that the characteristic signature is the result of proper removal of the cross-links from the damaged sites in the DNA. If this healing process is jeopardized or the repair machinery gets overloaded, instead gross chromosomal changes and chromosomal aberrations seem to occur when the damaged cells attempt to overcome the DNA cross-links. The evidence of such botched repair is frequent in colorectal cancers and suggests that the carcinogenic effect of colibactin may be substantially greater than the ten percent of cases suggested by the signature alone”.
###
Original publication
Iftekhar A, Berger H, Bouznad N, Heuberger J, Boccellato F, Dobrindt U, Hermeking H, Sigal M, Meyer TF.
Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells.
Nature Communications
February 12, 2021






