Computer scientists develop tool for mining genomic data
Equipped with cutting-edge techniques to track the activity of tens of thousands of genes in a single experiment, biologists now face a new challenge – determining how to analyze this tidal wave of data. Stanford Associate Professor of Computer Science Daphne Koller and her colleagues have come to the rescue with a strategic approach that reduces the trial-and-error aspect of genetic sequence analysis.
’’What we’re developing is a suite of computational tools that take reams of data and automatically extract a picture of what’s happening in the cell,’’ says Koller. ’’It tells you where to look for good biology.’’
Koller presented her statistical approach for mining genomic data at a Feb. 14 symposium – ’’Machine Learning in the Sciences’’ – at the annual meeting of the American Association for the Advancement of Science (AAAS) in Seattle.
Several years ago, before Koller came onto the scene, a new generation of high-throughput assays revolutionized molecular biology. In the most stunning example of this technology, scientists began using thumbnail-sized ’’gene chips’’ to monitor the activities of thousands of genes at once. In October 2003, Santa Clara-based Affymetrix took this breakthrough to a new level when it began marketing whole-genome chips packed with all 30,000 to 50,000 known human genes. Genome chips can reveal, for instance, that in kidney cells treated with a certain drug, 116 genes spring into action while another 255 get shut off.
But this state-of-the-art DNA microarray technology provides only a single snapshot of the cell. ’’It’s a very partial view,’’ Koller says.
What scientists really want to know is how groups of genes work together to control specific biological processes, such as muscle development or cancer progression. Unraveling these regulatory networks – for example, determining that Gene A gets activated by Gene B but repressed by Gene C – is a daunting task.
Sifting through whopping amounts of DNA microarray data to cull the hundreds of activator and repressor candidates is actually the easy part. The real challenge is figuring out which of these genes, if any, are biologically meaningful. This requires a bewildering array of hit-or-miss wet-lab experiments that examine protein-protein and protein-DNA interactions among the candidate genes.
Koller’s computational tools will make this scheme less formidable by providing scientists with targeted hypotheses in the form of ’’Gene A regulates Gene B under Condition C.’’ These predictions are generated from a probabilistic framework that integrates data from a variety of sources, including microarrays, DNA sequences, and protein-protein and protein-DNA interactions.
As Koller sees it, each of these sources offers a glimpse into what is happening in the cell: ’’a snapshot from this angle, a shot from another angle, data from a third, and so on.’’ Her computational scheme creates ’’the best picture we can construct from putting all of these snapshots together.’’
The proof of concept for Koller’s targeted hypotheses came in a June 2003 Nature Genetics publication, which described the application of her tools to predict gene regulatory networks in a variety of biological processes in yeast. Three of these predictions were confirmed in wet-lab experiments, suggesting regulatory roles for previously uncharacterized proteins.
’’The creativity and computer science perspective brought to these problems by Koller and her collaborators provide a tremendous boost to biology,’’ says Matthew Scott, a developmental biologist at Stanford and chair of the scientific leadership council of Bio-X, an interdisciplinary initiative. His research group has used Koller’s approach to identify genes involved in specific processes during embryonic development, to determine which genes are key regulators of other genes and to track changes in gene activities during disease progression.
Scott adds that while the computational methods suggest interesting hypotheses, their ultimate validation relies upon lab experiments.
In the future, Koller hopes to develop her scheme to handle multi-species analysis – for instance, to identify gene regulatory networks that appear in both human and mouse genomes. ’’When a regulatory module is conserved across multiple species, that indicates it’s playing a significant role,’’ Koller says.
Koller’s collaborators include Eran Segal and Michael Shapira (both of Stanford), Nir Friedman (Hebrew University of Jerusalem), Aviv Regev (Harvard Center for Genome Research), Dana Pe’er (Harvard-Lipper Center for Computational Genetics), Roman Yelensky (Massachusetts Institute of Technology) and David Botstein (Princeton University).
Media Contact
All latest news from the category: Information Technology
Here you can find a summary of innovations in the fields of information and data processing and up-to-date developments on IT equipment and hardware.
This area covers topics such as IT services, IT architectures, IT management and telecommunications.



Newest articles
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
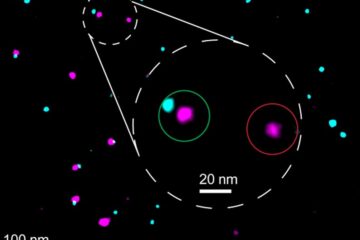
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

