Protein discovered that keeps hemoglobin in balance
Research at The Children’s Hospital of Philadelphia may advance treatment of the blood disease thalassemia
Hematology researchers at The Children’s Hospital of Philadelphia have discovered a gene and its associated protein that may have major implications for red blood cell formation, specifically for hemoglobin, which carries oxygen in red blood cells. Understanding how this protein functions may eventually lead to novel treatments for the hemoglobin-related blood disease, thalassemia.
Thalassemia is a group of related inherited disorders that together are the most common single-gene disease known. The most severe form of the disease, beta thalassemia major, affects 300,000 patients worldwide.
Thalassemia results from an imbalance between two proteins in hemoglobin, called alpha and beta globin. An excess of either type of protein is toxic, causing thalassemia symptoms including poor growth, fatigue, bone damage, or skin ulcers. The newly found protein, alpha hemoglobin stabilizing protein (AHSP), binds to free alpha globin and prevents it from forming a precipitate that damages red blood cells.
“AHSP acts as a chaperone molecule – a chemical that helps another protein to fold or unfold,” said Mitchell Weiss, M.D., Ph.D., a pediatric hematologist at The Children’s Hospital of Philadelphia, and senior author of the paper, published in the June 13 issue of Nature. “Here it makes free alpha globin stable and prevents its deleterious effects.”
Dr. Weiss’ team suspected that AHSP, by preventing free alpha hemoglobin from precipitating within red blood cells, protects the cells from injury. To test that hypothesis, the researchers developed knockout mice, animals genetically engineered to lack the gene that produces AHSP. Those mice showed blood abnormalities similar to those found in mice with thalassemia.
AHSP’s protective role could explain how some patients who carry the genetic trait for beta thalassemia have mild disease and few symptoms even though their bodies produce more alpha than beta globin. By binding to free alpha globin, AHSP may protect the body from a dangerous accumulation of that protein. However, if AHSP does not function properly, the excess alpha globin precipitation may change milder or intermediate thalassemia into more severe disease.
This current research suggests that if physicians can deliver AHSP or a similar agent to patients with thalassemia, they may produce a new treatment for the disease. Severe cases are now treated with frequent blood transfusions that carry their own serious complications, such as excess iron. “If we can reduce the buildup of free alpha globin we may be able to lower the dose of transfusion needed, and improve patients’ quality of life,” added Dr. Weiss.
In addition to Dr. Weiss, co-authors of the article are Anthony J. Kihm, Ph.D., Yi Kong, Wei Hong, Ph.D., J. Eric Russell, M.D., Susan Rouda, Kazuhiko Adachi, Ph.D., and Gerd A. Blobel, M.D., Ph. D., all of Children’s Hospital’s Division of Hematology, and M. Celeste Simon, Ph.D., of the Howard Hughes Medical Institute and the University of Pennsylvania. The research was funded by the Cooley’s Anemia Foundation, the Unico Foundation and the National Institutes of Health.
Founded in 1855 as the nation’s first pediatric hospital, The Children’s Hospital of Philadelphia is ranked today as the best pediatric hospital in the nation by a comprehensive Child magazine survey. Its pediatric research program is among the largest in the country, ranking second in National Institutes of Health funding.
Media Contact
All latest news from the category: Health and Medicine
This subject area encompasses research and studies in the field of human medicine.
Among the wide-ranging list of topics covered here are anesthesiology, anatomy, surgery, human genetics, hygiene and environmental medicine, internal medicine, neurology, pharmacology, physiology, urology and dental medicine.



Newest articles
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
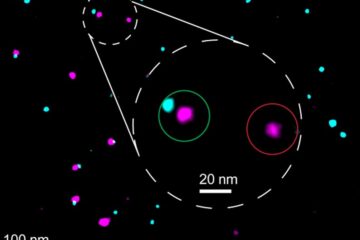
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

