‘Smart’ genetic therapy helps the body to heal itself
Professor Brunhilde Wirth, from the Institute of Human Genetics, University of Cologne, Germany, will describe to the conference her team’s work on developing therapies for patients with spinal muscular atrophy (SMA). This is a relatively common inherited disease in humans and the leading cause of death in infants, with an incidence of about 1 in every 6,000 newborns. Due to degeneration of the motor neurons in the spinal cord patients develop muscle weakness and atrophy of the legs, arms and trunk.
In patients with SMA the survival motor neuron genes (SMN1) is deleted, but they all carry a copy gene (SMN2). However, this only produces about 10% of the correct protein; insufficient to prevent the diseases. The severity of the SMA is influenced by the number of SMN2 genes, which normally vary between one and four – the more copies there are the better the patient does.
Professor Wirth’s team identified a drug – valproate, used successfully in epilepsy treatment for decades – that is able to increase SMN protein levels 2-4 fold in cell lines derived from SMA patients. Later, they showed that valproate also raised SMN levels in neuronal tissues such as cultured brain slices derived from epilepsy patients after surgery, as well as motor neuron cultures derived from rat embryos.
“The next step was to try this therapy in patients”, said Professor Wirth. “In a first pilot trial, we enrolled ten parents of SMA patients who were treated with valproate for four months. We saw that valproate significantly increased SMN levels in blood. Based on this data, 20 SMA patients were treated with valproate by their local doctors. 7 of these patients showed increased SMN2 levels in their blood. With the help of a biomarker we hope we will be able to distinguish between patients who will respond to this drug and which will not.”
The results show that valproate is able to exert a direct effect on the activity of the human SMN gene, says Professor Wirth, but it is still unclear whether SMN expression in blood reflects SMN expression in motor neurons, and hence will have an effect on muscle strength. “These pilot studies have to be followed up by Phase II and III clinical trials in SMA patients,” she said, “but unfortunately, there is very little interest from the pharmaceutical industry in clinical trials for rare disorders. However, the long-term outcome could be both improved therapy to enable a better quality of life for SMA patients, and also the introduction of neonatal screening so that therapy could be started before the first symptoms appear.”
Another study involving valproate will be presented by Professor Aurora Pujol, of the Medical and Molecular Genetics Centre- IDIBELL and the Institució Catalana de Recerca i Estudis Avançats (the Catalan Research Agency) in Barcelona, Spain. Dr. Pujol and her team are trying to develop new therapies for X-linked adrenoleukodystrophy (X-ALD). This disease involves a single gene on the X chromosome (the ALD gene) and is the most common inherited single-gene disease involving damage to the myelin sheath, which insulates nerve cells in the brain.
Using a mouse model of the disease, Dr Pujol and colleagues found that a protein called ALDR (very similar to the ALD protein mutated in the disease) could, if over-expressed, compensate for the loss of the ALD protein and prevent the development of neurodegenerative symptoms. This striking prevention of the disease lasted during the whole 2 year life span of the mouse.
“Knowing that a class of drug called histone deacetilase (HDAC) inhibitors was able to stimulate the expression of ALDR in the mouse and in human cell lines, we became interested by the work of Professor Wirth’s laboratory on valproate, which belongs to the HDAC class and enters the brain ”, said Professor Pujol.
The scientists tested valproate and found that it could indeed induce ALDR gene expression in human cell lines and rat and human brain slices. “We then treated 8 X-ALD patients with oral valproate over 6 months”, said Professor Pujol. “We collected samples before treatment, and at 3 and 6 months, and on analysing the amount of ALDR in their white blood cells, we found that half of them showed increased levels.”
The scientists now intend to measure the actual marker of the disease, the very-long chain fatty acids in white blood cells and plasma. If these levels have normalised with valproate, they hope to be able to start a double-blind clinical trial in collaboration with Professor Patrick Aubourg of INSERM, the French national medical research agency, at the Hospital Saint Vincent de Paul, Paris.
“Treatment with valproate could ameliorate symptoms or prevent neurodegeneration, especially in the late-onset type of the disease”, said Professor Pujol. “Currently there is no effective therapy available for this devastating disease, which can cause visual loss, seizures, speech problems or deafness due to demyelination invariably leading to death in children and problems of gait and co-ordination in adults.”
Professor Gert-Jan van Ommen, from the Centre for Human and Clinical Genetics, Leiden University Medical Centre, The Netherlands, and colleagues, will tell the conference that the team of Dr. Judith van Deutekom in his department has discovered a promising genetic therapy for Duchenne muscular dystrophy (DMD). Using anti-sense molecules (small pieces of synthetic RNA that can bind to specific sequences in the parts of the gene that code for the disease), the processing of the genetic code for dystrophin – a protein important for the muscle fibre membrane – can be manipulated in such a manner that certain units (‘exons’) are skipped. In this way, the readability of the code can be restored and the progress of DMD delayed or even stopped.
In collaboration with the Dutch biotech company Prosensa BV, and supported by the national and international Duchenne Parent Projects, and governmental and charity funds, Dr. J. van Deutekom and her team will soon start a clinical trial, based on intramuscular injection of a single dose of an anti-sense drug that induces the skipping of exon 51 from the DMD gene. Six Dutch DMD patients aged between 8 and 16 years with genetic mutations that are correctable by skipping that particular exon will take part.
This human trial is being supported by animal work. The team has used the muscular dystrophy mouse model to study optimal dose regimes, and further study exon skipping levels. This study found no clinical adverse signs, and the restoration of dystrophin levels, even in the heart.
“Of all the different strategies against DMD currently being contemplated,” said Professor van Ommen, “this is the one closest to clinical application. The patients in the trial have already been pre-screened, and we have seen the effect of the anti-sense drug in their isolated and cultured muscle cells. We are optimistic that this small scale trial will be the precursor of major clinical studies, which will lead in turn to a safe and efficient full body treatment for DMD patients. Currently no therapy is available for this disease, which represents a huge psychosocial and economic burden to patients, carers and society.”
Media Contact
More Information:
http://www.mrcommunication.orgAll latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
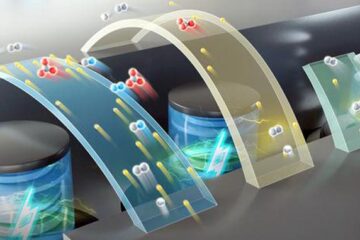
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
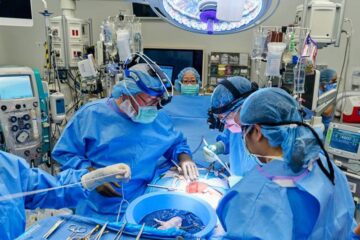
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

