Evolution in Action: Why Some Viruses Jump Species
Researchers studying strains of a lethal canine virus and a related human virus have determined why the canine virus was able to spread so quickly from cats to dogs, and then from sick dogs to healthy dogs. Their studies may lead to a new understanding of the critical molecular factors that permit viruses to jump from one species to another — information that could be helpful in assessing how much of a threat avian influenza is to humans.
In advance online publication of a paper in the April 2006 issue of the Journal of Virology, Laura Shackelton, an HHMI predoctoral fellow at the University of Oxford in England, examined the surprisingly rapid evolution of the B19 erythrovirus, a ubiquitous human parvovirus.
Shackelton’s latest paper extends her previous research published in the Proceedings of the National Academy of Sciences in 2005 on the carnivore parvoviruses, specifically panleukopenia virus (FPLV), a feline virus that crossed over into dogs over 30 years ago. The work was done in collaboration with her advisor, former Oxford professor Edward C. Holmes, who is the senior author on the papers. Holmes recently moved his laboratory to Pennsylvania State University, where Shackelton will join him for postdoctoral research.
Shackelton’s work can be considered a genomic analysis of evolution in action. “Viruses don’t leave fossils,” she explains. “But if you compare the differences between extant viral sequences, you can calibrate the molecular clock.”
Viruses penetrate the interior of cells by binding to receptors on the host cell’s surface. The binding occurs in much the same way that a key fits into a lock. Sometimes proteins on the outer coat of a virus mutate enough to match the receptors on the cells of species other than ones that the virus usually infects. This is what has happened with avian flu. Once in a new species, the virus either dies out or is preserved and adds mutations that enable it to move from host to host within the new species. This can lead to widespread infection.
Shackelton wanted to understand the process of host-switching, the molecular mechanisms viruses employ to jump from one species to another. “We found the carnivore parvoviruses to be an excellent model for studying the molecular changes that accompany host-switching,” she said, “because it’s one of the very few viruses for which we have adequate sequence data before and after the cross-species transfer.”
Colin R. Parrish, a parvovirus expert at Cornell University, and Uwe Truyen of the University of Leipzig, co-authors on the PNAS paper, provided the genomic sequences of parvovirus stocks that dated back to the 1960s. Working with the sequences, Shackelton traced backwards to construct a phylogenetic tree showing when mutations in the virus became fixed and started being transmitted from generation to generation.
In order to estimate the evolutionary dynamics of the virus, she and Holmes used a Bayesian Markov Chain-Monte Carlo (MCMC) approach. A powerful statistical tool, the Bayesian MCMC method looks at probabilistic relationships. “This approach allows us to utilize the information in the viral sequences, and their isolation dates, to thoroughly explore models and modes of their molecular evolution,” says Shackelton.
Their analysis showed that the version of the virus found in cats had been infecting the feline population for over one hundred years. But once it began to infect canines, FPLV, now canine parvovirus (CPV-2), quickly accumulated additional nucleotide substitutions, or changes in individual DNA building blocks, and gained the ability to transfer from an infected dog to a healthy dog. The rapid pace of that change contradicted conventional wisdom, which says that DNA viruses do not mutate rapidly.
Parvoviruses are single-stranded DNA viruses that package one strand of DNA in a protective shell, called a protein capsid. Once in the host cell, this strand acts as the template, and the virus replicates through a double-stranded intermediate. The more common double-stranded DNA viruses need to have the coiled helix unwind in order to replicate.
“Since they’re DNA viruses and replicate with host-cell machinery, it was just assumed that we should see rates of mutation more on par with those of their hosts and other DNA viruses,” said Shackelton. “Instead, we were seeing orders-of-magnitude differences, rates more characteristic of RNA viruses.”
Intrigued, she then set out to see if this phenomenon was characteristic of the entire Parvoviridae family. In the research reported in the Journal of Virology, she examined the human B19 erythrovirus, a parvovirus that infects bone marrow progenitor cells and can be associated with heart complications. She chose it because it is distantly related to CPV-2, with different routes of infection. The analysis showed the same surprisingly rapid rate of change. She and Holmes are now confident that “the difference between DNA and RNA viruses is not as simple as we thought it was. There’s something unique about parvoviruses that possibly extends to all single-strand viruses.”
In their next series of experiments, together with Parrish’s group, the scientists will work to understand more about that uniqueness and learn its underlying cause. It is work that may help in understanding all viruses, including avian flu.
“The more we know about species transfer events and factors influencing nucleotide substitution rates, the better,” says Shackelton, who is also interested in public policy responses to viral outbreaks, “This work is increasing our knowledge about what molecular changes need to happen for species transfer, which changes or combinations of changes are the critical ones, and how frequently they occur.”
Media Contact
More Information:
http://www.hhmi.orgAll latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
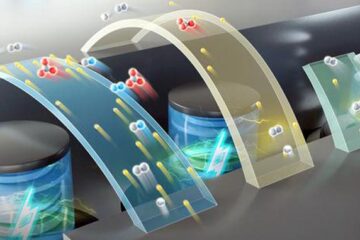
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
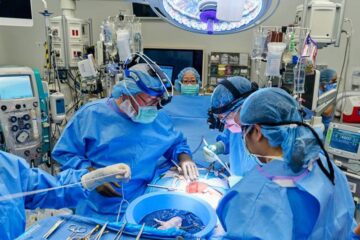
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

