Blocking a premature aging syndrome with anticancer drugs
A class of anticancer drugs currently being evaluated in phase 3 clinical trials may also be an effective treatment for Hutchinson-Gilford progeria syndrome (HGPS), a fatal genetic disorder that causes premature aging. If upcoming studies in a HGPS mouse model validate the results of experiments in cultured cells in the laboratory, a clinical trial of these drugs in HGPS children may begin as early as next spring.
Brian Capell, a New York University medical student participating in the Howard Hughes Medical Institute/National Institutes of Health Research Scholars Program and the first author of the study, reported the findings in the August 29, 2005, issue of the Proceedings of the National Academy of Sciences. The HHMI-NIH Research Scholars Program gives outstanding medical and dental students the opportunity to conduct biomedical research under the direct mentorship of senior NIH research scientists.
Although HGPS is a rare disease that affects only one child in 4 million, the disease has received wide publicity due to its striking nature. Children born with HGPS appear normal, but they experience growth retardation and show symptoms of accelerated aging — namely hair loss, skin wrinkling, and fat loss — around the first year of age. Accelerated cardiovascular disease also ensues, which typically causes death around age 12.
In 2003, researchers in Francis Collins’s lab at the National Human Genome Research Institute discovered that mutations in the lamin A (LMNA) gene cause HGPS. The discovery has prompted renewed interest among researchers to study this rare syndrome.
When Capell entered Collins’s lab in July 2004, he immediately set his sights on understanding the molecular basis of HGPS. “What really interested me in this research in the first place were the potential links to aging and atherosclerotic disease,” says Capell. Indeed, understanding HGPS at the molecular level may illuminate general processes involved normal human aging.
The LMNA mutation implicated in HGPS causes an internal stretch of 50 amino acids within the encoded lamin A protein to be deleted. This mutated protein is called “progerin.” Lamin A normally constitutes a major component of the scaffold-like network of proteins just inside the nuclear membrane called the lamina. When mutated to progerin, however, lamin A fails to integrate properly into the lamina, thereby disrupting the nuclear scaffolding and causing gross disfigurement of the nucleus. Cells with progerin have a nucleus with a characteristic “blebbed,” or lobular, shape.
To find its way to the lamina, lamin A carries two tags, rather like zip codes, that help to direct its travels. One tag at the end of lamin A instructs another protein to modify it through a process called farnesylation. Farnesylation tethers lamin A to the inner nuclear membrane. Once there, a second tag within the protein signals an enzyme to cleave off the terminal portion of the protein, including the farnesyl group, freeing lamin A to integrate properly into the nuclear lamina.
Because progerin carries the farnesylation tag but lacks the second cleavage tag, Capell speculated that progerin was becoming permanently stuck to the inner nuclear membrane. There, he suspected, it enmeshed other scaffolding proteins, preventing their proper integration into the lamina. If progerin’s sticking to the inner nuclear membrane is indeed the culprit in nuclear blebbing and the root of the HGPS defect, Capell reasoned that he could prevent these defects by blocking farnesylation of progerin.
Capell’s hunch proved correct. When he changed one amino acid within progerin’s farnesylation tag to prevent the addition of a farnesyl group and tested the effect in cells grown in the laboratory, progerin did not anchor itself to the inner nuclear membrane and instead clumped within the nucleus. Moreover, Capell observed no nuclear blebbing.
Capell then tried treating the cells carrying progerin with farnesyltransferase inhibitors (FTIs), drugs originally developed to inhibit certain cancer-causing proteins that require farnesylation for function, like the famous oncoprotein Ras, which are now being tested in phase III clinical trials of patients with myeloid leukemia. Again, he witnessed no blebbing. More importantly, he saw the same effect when he treated cells grown from skin biopsies of HGPS patients: HGPS cell blebbing decreased to near normal levels.
“FTIs, originally developed for cancer, are capable of reversing the dramatic nuclear structure abnormalities that are the hallmark of cells from children with progeria. This is a stunning surprise, rather like finding out that the key to your house also works in the ignition of your car,” says Collins, the principal investigator.
Capell’s work on progerin has so inspired him that he has decided to spend another year in Collins’s lab, with his next task being to test the effects of FTIs in their mouse model of HGPS.
Media Contact
More Information:
http://www.hhmi.orgAll latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
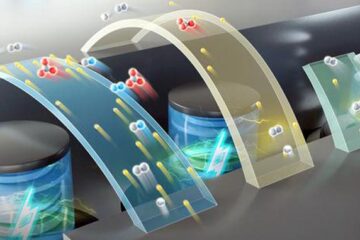
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
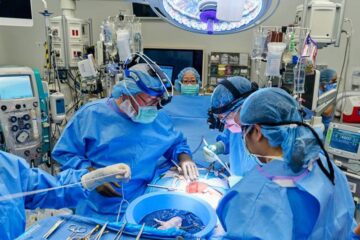
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

