New algorithm offers fast and accurate X-ray crystal structure identification
Identifying the structures of certain types of molecular compounds can now take minutes, instead of days, and be performed much more accurately, say scientists who developed a new approach for analyzing key experimental X-ray data.
Knowing the structure of a molecule allows scientists to predict its properties and behavior. While X-ray diffraction measurements have become a powerful tool for determining molecular structure, identifying the three-dimensional structure that best fits the diffraction data can be a major challenge.
As will be reported in the September issue of Acta Crystallographica Section A, researchers at the University of Illinois at Urbana-Champaign have developed an algorithm that provides fast and accurate structure determination for organic compounds and other molecular structures that have a center of symmetry.
In X-ray diffraction, a crystallized version of the target compound is bombarded by a beam of X-rays. Recorded by an X-ray detector, the two-dimensional patterns of diffracted wave intensities can be used to reconstruct the three-dimensional object.
“A big problem, however, is identifying the phases of the diffracted X-rays from measurements of intensities alone,” said Nikolaos Sahinidis, an Illinois professor of chemical and biomolecular engineering. “You know how strong the waves are, but you don’t know their phases, which are needed in order to compute the three-dimensional structure. This is known as the ’phase problem’ in crystallography.”
Crystallographers usually rely upon various trial-and-error methods to search for a solution that solves the phase problem and identifies the crystal structure. But such methods are time-consuming and do not guarantee a correct solution.
“Most methods for solving the phase problem make use of a merit function to score potential structures based on how well they match the experimental data,” Sahinidis said. “In the past, local optimization techniques and advanced computer architectures have been used to solve this problem, which may have a very large number of local optima.”
Sahinidis and graduate student Anastasia Vaia developed a new approach: reformulating the problem for the case of centrosymmetric crystal structure into an integer programming problem in terms of the missing phases.
“Integer programming problems have been studied extensively in the optimization literature,” Sahinidis said. “A great variety of combinatorial optimization methods have been developed to solve these problems without explicitly trying all possible combinations of the missing phases.”
By introducing integer programming into crystallographic computing, “we can use off-the-shelf optimization software to rapidly find the correct solution to the phase problem,” Sahinidis said. “We were able to solve many X-ray structures for which popular crystallographic software failed to provide a solution. No trial-and-error is required by our algorithm and there is no ambiguity that the correct three-dimensional structure has been identified.”
Sahinidis and Vaia are now working to extend the integer programming approach to the more general case of non-centrosymmetric structures, which includes most proteins.
###
The University of Illinois, National Science Foundation and ExxonMobil Upstream Research Company funded the work.
Media Contact
More Information:
http://www.uiuc.edu/All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
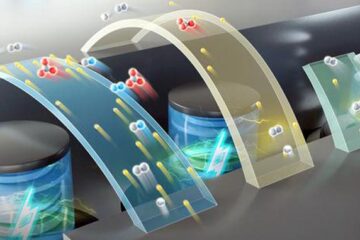
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
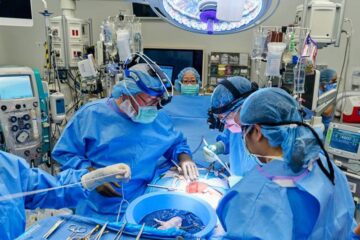
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

