Ray of hope for human Usher syndrome patients
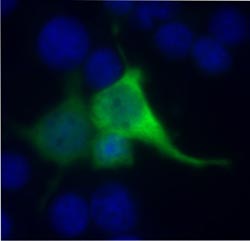
Fluorescence microscope image of cells with a nonsense mutation in the Usher syndrome 1C gene (USH1C/harmonin). In the presence of the designer aminoglycoside NB54, there is read-through of the USH1C mutation. The treated cells produce the healthy harmonin protein (green). Source: Kerstin Nagel-Wolfrum, Institute of Zoology, JGU<br>
After years of basic research, scientists at Johannes Gutenberg University Mainz (JGU) are increasingly able to understand the mechanisms underlying the human Usher syndrome and are coming ever closer to finding a successful treatment approach.
The scientists in the Usher research group of Professor Dr. Uwe Wolfrum are evaluating two different strategies. These involve either the repair of mutated genes or the deactivation of the genetic defects using agents. Based on results obtained to date, both options seem promising. Usher syndrome is a congenital disorder that causes the loss of both hearing and vision.
Usher syndrome is the most common form of congenital deaf-blindness in humans, occurring in 1 in 6,000 of the population. Those suffering from the disease are drastically handicapped in everyday life as they lose the use of the two most important sensory organs, i.e., their ears and eyes. In the most severe cases, patients are born deaf and begin to suffer from vision impairment in the form of retinal degeneration in puberty that result in complete blindness.
While it is possible to compensate for the loss of hearing with hearing aids and cochlear implants, no therapy was previously available for the ophthalmic component of the disorder. Scientists at Mainz University are currently undertaking preclinical translational research in an attempt to find an answer to this problem.
The investigations undertaken by the team of Dr. Kerstin Nagel-Wolfrum focused on the nonsense mutation in the USH1C gene that had been identified as the cause of the most severe form of Usher syndrome in a German family. The nonsense mutation is a stop signal generated by the DNA that causes premature termination of synthesis of the protein harmonin, which is encoded by USH1C.
The research team published its latest findings with regard to gene repair as a possible treatment of Usher syndrome in the June edition of the opthalmologic journal Investigative Opthalmology & Visual Science. During her doctoral research, Dr. Nora Overlack managed to repair the USH1C gene with the help of molecular scissors' generated using the so-called zinc-finger nuclease technique. Using zinc-finger nuclease, the scientists first initiated a double sequence DNA cleavage at the site of the disease-generating mutation.
This surgical incision on the molecular level was then repaired by means of the cell's own repair mechanism in the form of homologous recombination and the introduction of a non-mutated USH1C DNA sequence. The mutated gene sequence was thus replaced with the non-mutated sequence. The efficacy of the zinc-finger nuclease technique with regard to genetic repair was demonstrated in a cell culture model at both the genome and the protein level.
The research team has also recently published the latest results of its pharmaco-genetic approach to the treatment of Usher syndrome patients with nonsense mutations in the journal EMBO Molecular Medicine. In this case, Dr. Tobias Goldman and the other team members compared various molecules that can induce read-through of the stop signal and thus provide for normal protein synthesis. In addition, they evaluated the retinal biocompatibility of the various molecules. The research focused on PTC124 (Ataluren®) and 'designer' aminoglycosides. These aminoglycosides are derived from clinically tested antibiotics and have been modified by Professor Dr. Timor Bassov of the Technicon in Haifa/Israel to improve their capacity to read-through the mutation and reduce their toxicity. The Mainz researchers had already been successful in using one of the first generation designer aminoglycosides to read-through the nonsense mutations in the USH1C gene.
They were now able to show that PTC124 (Ataluren®) and a second generation aminoglycoside (NB54) in particular would induce read-through of the stop signal in the mutated USH1C gene. This meant that protein synthesis continued, so that the active gene product was synthesized in the cell and organ cultures. Both active substances, PTC124 and NB54, generally enhanced read-through efficacy and exhibited improved tolerability in mouse and human retinal cultures in comparison with clinically employed antibiotics. The team also successfully documented read-through of the mutation in vivo a mouse model.
“Our gene-based treatment strategies, involving gene repair as well as read-through therapy, represent valuable and promising alternatives to viral gene addition and may actually be the only treatment option for the large and isoform-rich USH genes. We hope that these alternatives will make a significant contribution to the therapy of both Usher syndrome patients as well as others with severe genetic retinal pathologies and other genetic disorders,” explains Dr. Kerstin Nagel-Wolfrum.
In addition to continuing its preclinical studies into the use of the active substances, the Mainz Usher research team plans to make its new Usher syndrome therapy available to patients as soon as possible.
The translational biomedical research into the treatment of Usher syndrome was carried out with the help of financial support from the EU-FP7 project SYSCILIA, the FAUN foundation, and the Foundation Fighting Blindness (FFB). The two involved doctoral candidates were research assistants and colleagues in the Research Training Group 1044: “Developmental and disease-induced modifications of the nervous system” supported by the German Research Foundation. The work of the Usher syndrome researchers is integrated in the Research Unit Translational Neurosciences (FTN) at Johannes Gutenberg University Mainz.
Publications:
Nora Overlack, Tobias Goldmann, Uwe Wolfrum, Kerstin Nagel-Wolfrum
Gene repair of an Usher syndrome causing mutation by zinc-finger nuclease mediated homologous recombination
Investigative Ophthalmology & Visual Science, June 2012
doi:10.1167/iovs.12-9812
Tobias Goldmann et al.
A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation
EMBO Molecular Medicine, October 2012
doi:10.1002/emmm.201201438
Further information:
Dr. Kerstin Nagel-Wolfrum
Cell and Matrix Biology
Institute of Zoology
Johannes Gutenberg University Mainz (JGU)
D 55099 Mainz, GERMANY
phone +49 6131 39-20131 or 39-23934
fax +49 6131 39-23815
e-mail: nagelwol@uni-mainz.de
http://www.ag-wolfrum.bio.uni-mainz.de/
Media Contact
All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.




Newest articles

A universal framework for spatial biology
SpatialData is a freely accessible tool to unify and integrate data from different omics technologies accounting for spatial information, which can provide holistic insights into health and disease. Biological processes…

How complex biological processes arise
A $20 million grant from the U.S. National Science Foundation (NSF) will support the establishment and operation of the National Synthesis Center for Emergence in the Molecular and Cellular Sciences (NCEMS) at…

Airborne single-photon lidar system achieves high-resolution 3D imaging
Compact, low-power system opens doors for photon-efficient drone and satellite-based environmental monitoring and mapping. Researchers have developed a compact and lightweight single-photon airborne lidar system that can acquire high-resolution 3D…

