Simulations on biologically relevant time scales
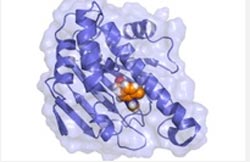
In order to achieve the simplification of system dynamics, physicists have developed the dissipation-corrected targeted Molecular dynamics simulations. Credit: Steffen Wolf/AG Gerhard Stock
Molecular dynamics simulations (MD) have become an ubiquitous tool in modern life sciences. In these simulations, the interactions between atoms and molecules and their resulting spatial movements are iteratively calculated and analyzed. Scientists are currently trying to gain access to biologically relevant length and time scales using this approach in order to describe molecular processes such as protein folding and protein-drug binding, which are crucial for, for example modern drug development.
A team led by Dr. Steffen Wolf and Prof. Dr. Gerhard Stock from the Biomolecular Dynamics group at the Institute of Physics of the University of Freiburg has now succeeded in predicting the dynamics of binding and unbinding processes on a time scale of seconds to half a minute in pharmacologically relevant test systems. The results have been presented in the current issue of the journal Nature Communications.
Due to the need to perform atomistic simulations with a temporal resolution of femtoseconds (10-15 s), researchers are not yet able to explicitly calculate processes that take a few or more seconds, such as the binding and release of drugs to and from their respective target protein.
One possible approach to speed up simulations is coarse-graining of the overall system dynamics, which is a domain of non-equilibrium statistical mechanics. To achieve this coarse-graining, slow processes such as protein-ligand diffusion and fast processes like protein vibrations or water fluctuations must exhibit a clear time scale separation.
Only then can scientists use the Langevin equation, a stochastic differential equation that describes the dynamics along the relevant slow degrees of freedom – that is, the number of independent possibilities of movement — of a physical system. Using this equation, they represent the dynamics of the system along a reaction coordinate such as the distance of the ligand from its binding site. All other, faster movements are considered as friction.
In order to achieve this necessary simplification of system dynamics, the Freiburg physicists have developed the dissipation-corrected targeted MD (dcTMD) using computational resources from the HPC cluster BinAC at the University of Tübingen.
By applying a constraining force to actively pull a microscopic system along a coordinate of interest, the required work can be broken down into free energy and friction fields of the process.
In the current publication, the researchers have shown that these dcTMD fields can be used as input for a simulation of the Langevin equation along the pulling coordinate. As a result, the researchers have been able to greatly reduce the required computational power. A simulation time of one millisecond can thus be achieved within a few hours on a single computing core of a standard desktop computer.
In addition, Langevin fields, explains Stock, do not change their structure at higher temperatures, unlike to atomistically described proteins. “Therefore, high-temperature simulations can produce accelerated dynamics. We can use this acceleration to extrapolate the dynamics at a lower temperature of interest, where the fields are derived from targeted MD simulations.”
The Freiburg scientists used the dissociation of sodium chloride and two protein-ligand complexes as test systems. In these they succeeded in predicting the dynamics of binding and unbinding processes on a time scale of seconds to half a minute.
“While the Langevin fields were only generated from unbinding simulations, they were able to predict both unbinding and binding kinetics within a factor 20 and dissociation constants within a factor 4, which is within the best achievable results compared to other prediction methods,” explains Wolf. At the same time, the new dcTMD approach requires only one tenth of the computing power of other prediction methods.
“Last but not least, the determination of friction profiles provides insights into molecular processes that are not revealed by free energy,” say the Freiburg physicists. “We found that in all the systems investigated, the formation of a hydration shell from water molecules seems to be the main source of friction. This enables us to deduce new rules for the design of drugs with desired binding and diffusion kinetics.”
Original publication:
Wolf, S., Lickert, B., Bray, S., Stock, G. (2020): Multisecond ligand dissociation dynamics from atomistic simulations. In: Nature Communications. DOI: 10.1038/s41467-020-16655-1
Contact:
Dr. Steffen Wolf and Prof. Dr. Gerhard Stock
Institute of Physics
University of Freiburg
Tel.: 0761/203-5913
E-Mail: steffen.wolf@physik.uni-freiburg.de; gerhard.stock@physik.uni-freiburg.de
https://www.pr.uni-freiburg.de/pm-en/press-releases-2020/simulations-on-biologic…
Media Contact
All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
Microscopic basis of a new form of quantum magnetism
Not all magnets are the same. When we think of magnetism, we often think of magnets that stick to a refrigerator’s door. For these types of magnets, the electronic interactions…

An epigenome editing toolkit to dissect the mechanisms of gene regulation
A study from the Hackett group at EMBL Rome led to the development of a powerful epigenetic editing technology, which unlocks the ability to precisely program chromatin modifications. Understanding how…

NASA selects UF mission to better track the Earth’s water and ice
NASA has selected a team of University of Florida aerospace engineers to pursue a groundbreaking $12 million mission aimed at improving the way we track changes in Earth’s structures, such…

