Study analyzes dynamical properties in antibiotic resistance enzyme
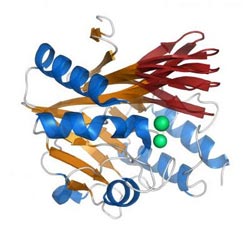
This is beta-lactamase, showing different permutations at the active site.<br><br>Credit: Dennis Livesay<br>
Antibiotic-resistant bacteria have been emerging at an alarming rate. In some of the scariest of these pathogens, the mechanism responsible for the bacteria's ability to defeat antibiotics is a complex protein molecule embedded in the bacterial cell wall — the enzyme â-lactamase.
The rapid evolution of â-lactamase is the key factor responsible for the growing antibiotic resistance of some of the most terrifying pathogenic bacteria on the planet – bacteria which are becoming rapidly immune to most, if not all, of our drugs. We can trace the genetic changes responsible, but actually understanding what those changes are doing to the properties of the hugely complex molecule is another matter.
The enzyme and its antibiotic-destroying effects are not new. â-lactamase has evolved over the millennia as a defensive weapon, a molecular machine for chopping up chemical weapons deployed in the wars bacteria fight against each other – antibacterial weapons that we have since discovered and call “antibiotics.” Because this chemical warfare has gone on for billions of years, the protein can be found in subtle variants in many bacteria.
Some of the variations in â-lactamase are ancient, but some are very recent, as the molecule has experienced intense evolutionary pressure in the last century due to human over-use of antibiotic compounds. However, it is still somewhat of mystery what specific structural or chemical changes in the protein have allowed its recent rapid changes in counter-antibiotic capability.
Now, new research appears to have uncovered the mechanisms involved, both in the protein's long-term evolution and in the specific changes responsible for the rapid development of resistance against antibiotics. In a finding published July 18 in the online edition of PLOS Computational Biology, University of North Carolina at Charlotte researchers Dennis R. Livesay, Deeptak Verma, and Donald J. Jacobs show significant evolution in the structural characteristics and physiochemical properties of â-lactamase across bacterial families, but also find that these evolutionary characteristics do not appear to be specifically related to different versions of antibiotic resistance.
Instead, the researchers found that relatively minor changes in the structure of the enzyme's active site – the area of the protein that couples with the antibiotic molecules and disables them – are capable, independent of global changes to the protein structure, of adapting the enzyme to new antibiotics. Though it was not the result the researchers were hoping to discover, the finding does have large implications.
“It's actually a cautionary result because it highlights that these mutations are not being restricted too much by the global properties of the enzyme,” said Livesay, a faculty member in bioinformatics. Livesay notes that different families of bacteria have evolved significant physiochemical differences in their â-lactamase molecules, but that these structural differences have allowed resistance to the same medically administered antibiotics to develop nonetheless.
Livesay's team studied the structure and properties of class-A â-lactamase proteins, one of four “families” of the protein that have evolved in bacteria and other organisms. They analyzed about a dozen proteins – those in the group whose structures have previously been described – and defined the intricate physiochemical properties of each of the proteins' structures, while comparing the individual protein structure characteristics they discovered with the phylogenetic trees of the bacteria they came from.
Central to their approach was the Distance Constraint Model (DCM), a program developed by Jacobs, a UNC Charlotte physicist, and Livesay. The DCM allows detailed but also relatively fast analysis of the protein structure's physical properties. The DCM's efficient but accurate structural analysis allowed the researchers to make complex structural comparisons between many different (but related) molecules – an analysis that would otherwise require vast amounts of processing power. The analysis allowed the researchers to pinpoint specific differences between the proteins, such as differing amounts of rigidity/flexibility in specific parts of the protein's complex structure.
“Biology is an inherently comparative science. From Darwin's finches to modern molecular biology, we frequently learn most through comparisons. In this work, we extend the comparison paradigm to computational biophysics by leveraging the speed and accuracy of the DCM.” Livesay said.
'We started by asking a very simple question: do the physical and chemical properties vary in a way that directly reflects the divergence of the family?” Livesay said. “What we did was calculate these properties and ask if those in the same evolutionary outgroups have similar properties and are those in different outgroups likely to have different properties?”
“We did some simple calculations and we proved conclusively that the physiochemical properties are varying in statistically significant way with the phylogeny. This is really cool,” he noted, “because it demonstrates that evolution is manipulating chemistry in a straightforward way.”
The next step was for the researchers to compare the genetically linked structural properties of the proteins to different varieties of antibiotic resistance in the bacteria. Livesay notes that antibiotic resistance in bacteria has long been studied and, in fact, used as an alternative form of classification.
“We wanted to see if we could link the properties we calculate to these activities. And it turns out, No, we can't,” Livesay said. “Frankly, I was a little disappointed when we first saw that. What's happening is that within a lineage the global properties change very little, but the severity of their response to antibiotics can be huge.”
Though the properties of the protein vary from one bacterial family to another, the researchers concluded that the entire â-lactamase group has general characteristics that prevent the protein's basic physiochemical properties from affecting the enzyme active site, where antibiotics are attacked.
“This enzyme is a rock,” Livesay noted. “It's atypically rigid — much more rigid than most proteins. So how does the enzyme become active against an antibiotic it wasn't active against before? Well it had some chemical groups that were simply in the way, meaning steric clashes would restrict what antibiotics could fit in the active site of the enzyme.”
The implication is that the general rigidity of â-lactamase allows relatively simple genetic changes – changes affecting only the structure of the active site – to cause new antibiotic resistance without otherwise affecting the behavior of the protein.
“It doesn't require any wholesale change in the protein's global properties to manipulate this local chemistry, so it turns out that these mutations are evolutionarily cheap,” Livesay said. “You can evolve these slight changes in the active site against any background of the global properties. A very small number of gene changes and a very small number of amino acid changes in the protein are involved.”
Since the larger structure of the protein does not interact with the behavior of the active site, Livesay stresses, it means that the same kinds of antibiotic resistance can re-occur across a broad range of bacteria from different families, though they have evolved differences in â-lactamase structure.
The ease with which the enzyme can evolve and adapt to new antibiotics, combined with the fact that some bacteria carry the â-lactamase gene on a plasmid (a separate ring of genetic material) that can be swapped with even unrelated bacteria, and the huge selective pressure caused by human overuse of antibiotics, all combine to create our current nightmare of widespread, rapidly developing antibiotic resistance.
According to Livesay, the current finding thus has larger implications.
“Our findings on class A â-lactamases are actually a much more terrifying result than one might expect,” he said. “It highlights that not only are their genes on mobile elements that are being transferred and shared, it turns out that these mutations are not being constricted too much by the physiochemical properties of the enzyme. That is presumably also contributing to the fact that â-lactamases in general have adapted so quickly.”
Going forward, the team is currently analyzing the â-lactamase genes that lead to carbapenem-resistant Enterobacteriaceae (CRE) infections. “Class B â-lactamases are the most dire, the most scary,” Livesay said. “These genes are highly mobile, on mobile elements and they are plastic and very active. They can be resistant to almost all the antibiotics we have. The enzyme can recognize in the active site all these different things, under different pH's. It's extraordinarily promiscuous. As such, CRE infections are very difficult to treat, leading to mortality estimates as high as 50% for infections that used to be treated with penicillin.”
This research is funded under grant number GM101570 from the National Institutes of Health. The complete paper can be seen online at http://www.ploscompbiol.org .
Source: Dennis Livesay, 704-687-7995, drlivesa@uncc.edu
Media Contact
More Information:
http://www.uncc.eduAll latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles

Why getting in touch with our ‘gerbil brain’ could help machines listen better
Macquarie University researchers have debunked a 75-year-old theory about how humans determine where sounds are coming from, and it could unlock the secret to creating a next generation of more…

Attosecond core-level spectroscopy reveals real-time molecular dynamics
Chemical reactions are complex mechanisms. Many different dynamical processes are involved, affecting both the electrons and the nucleus of the present atoms. Very often the strongly coupled electron and nuclear…
Free-forming organelles help plants adapt to climate change
Scientists uncover how plants “see” shades of light, temperature. Plants’ ability to sense light and temperature, and their ability to adapt to climate change, hinges on free-forming structures in their…

