In 15 seconds, new test will ID presence of 25 most common mutations of cystic fibrosis
“Eighty percent of all babies born in the US that have cystic fibrosis (CF) are born to parents with no previous family history.” This attention-getting quote, used frequently by an activist in the CF community, makes it powerfully clear what few people realize: that both parents did not know they would pass on to their child the life-altering CFTR gene, the gene mutated in cystic fibrosis.
In fact, the Cystic Fibrosis Foundation estimates that more than 10 million Americans are unknowing, asymptomatic carriers of CF, which causes a thickening mucous to surround the lungs and serves as a catalyst for multiple, life-threatening infections throughout a lifetime.
Background
Approximately 30,000 children and adults in the US suffer from CF. In the Caucasian population, where it is most prevalent, one in 30 white Americans carry the mutated gene in their genome but do not manifest the disease. When one carrier with a certain disease allele mates with another who carries the identical disease allele, a process of recombination produces an offspring with the double mutation necessary to cause the disease. One in 3,000 live births has CF today.
CF has more than 1,000 currently known mutations and additional mutations are being discovered regularly. Most of the mutations are “private,” running only in families and not outside a specific group. When the mutations exist within the family, many members will have it, the reason why so few common mutations exist.
The American College of Obstetrics and Gynecology (ACOG), American College of Medical Geneticists (ACMG) and National Institutes of Health (NIH) last year recommended that all individuals considering conception be screened for the presence of a mutated CF gene. The group examined all known mutations and identified those most prevalent for each ethnic group, and have recommended screening for the 25 which are most likely to occur (the “ACOG 25”). Each of the 25 mutations has also been ranked according to a disease severity index. For example, the severity of R117H, a rare ACOG 25 CF mutation, is associated with the presence of 5T, a variation on another region of the CF gene. Test results often show this 5T variation alone. Such results may be misleading for physicians and patients because this particular variant is not indicative of CF unless associated with the rare mutation (R117H) detected in the ACOG panel. Therefore, a reflex panel has been recommended to test for this particular variation only in the presence of the mutation.
The ACOG guidelines require that the female’s genotype be evaluated first. In the event her DNA reveals she has a CF mutation, a genetic counselor will explain the severity of CF any offspring would have, should an identical mutation be found in the male. When both parties have the same mutation, the odds are one-in-four that the offspring will be CF-positive.
Genetic Testing for Cystic Fibrosis
To discuss the current state of CF testing technology is Cindy WalkerPeach, Ph.D, of Ambion Diagnostics, a division of Ambion, Inc. in Austin, TX. Dr. WalkerPeach will make her presentation entitled, “Cystic Fibrosis Testing: Current Platforms and Emerging Technologies,” during the 55th Annual Meeting of the American Association for Clinical Chemistry (AACC) in Philadelphia, PA, July 20-24, 2003.
The demand for testing has risen in the past year in response to the ACOG recommendations. Several manufacturers have developed screening tests to meet the increasing needs. Others are working on a newer generation of tests, classified as emerging technologies. Large laboratories have also conducted their own research and created in-house tests.
A New 15 Second Test
Ambion Diagnostics has developed a bead-array test that will become available in first quarter, 2004. The DNA test panel evaluates 31 CF mutations, including the 25 recommended for carrier screening by ACOG and ACMG. The test is divided into 2 parts, one for the ACOG 25 and if required, a separate reflex panel is performed to confirm the severity of detected mutation(s).
The bead-array test requires only that genomic DNA be taken from the patient, usually through a blood draw. Following the blood draw, the genomic DNA is isolated and single-tube PCR amplification of the genomic DNA is performed against the 31 different mutations. In one single reaction, the PCR products are hybridized to probes tethered onto small color-coded microspheres. Each microsphere is run through a laser beam that identifies the presence of a mutation. After hybridization, all ACOG 25 mutations are screened in 15 seconds.
A benefit of this test is allowing laboratories to run thousands of tests each week. With the dramatic increase in the desire for testing, following the ACOG recommendations, labs are experiencing delays in conducting their current tests and sending results to physicians. Another benefit of this test is that it can be easily expanded to test for selected CF mutations not included in the ACOG 25.
Conclusions
By knowing the family-specific mutations, other family members could be tested to identify CF carriers. However, since so many rare mutations have been described, not all mutations will be detected by the screening test. If the history or specific mutation is not known, a negative screening test will reduce, but not eliminate, the risk of being a carrier. The new test will allow potential parents to know more quickly if they are carriers of any of the ACOG 25 CF mutations. The hope is that for parents-to-be, such knowledge will not only help them to make more informed decisions, but also allow for treatment options that can greatly impact quality of life.
Media Contact
More Information:
http://www.aacc.orgAll latest news from the category: Health and Medicine
This subject area encompasses research and studies in the field of human medicine.
Among the wide-ranging list of topics covered here are anesthesiology, anatomy, surgery, human genetics, hygiene and environmental medicine, internal medicine, neurology, pharmacology, physiology, urology and dental medicine.



Newest articles

Why getting in touch with our ‘gerbil brain’ could help machines listen better
Macquarie University researchers have debunked a 75-year-old theory about how humans determine where sounds are coming from, and it could unlock the secret to creating a next generation of more…

Attosecond core-level spectroscopy reveals real-time molecular dynamics
Chemical reactions are complex mechanisms. Many different dynamical processes are involved, affecting both the electrons and the nucleus of the present atoms. Very often the strongly coupled electron and nuclear…
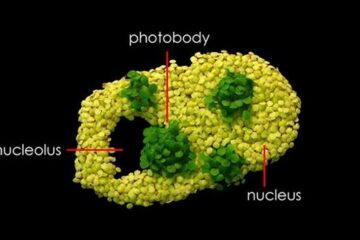
Free-forming organelles help plants adapt to climate change
Scientists uncover how plants “see” shades of light, temperature. Plants’ ability to sense light and temperature, and their ability to adapt to climate change, hinges on free-forming structures in their…

