Cell membrane proteins give up their secrets
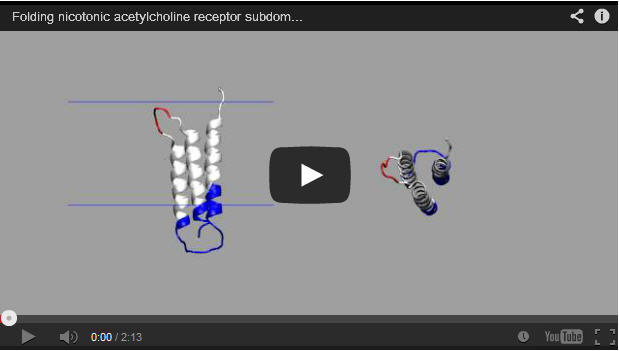
Rice University scientists have succeeded in analyzing transmembrane protein folding in the same way they study the proteins’ free-floating, globular cousins.
Rice theoretical biologist Peter Wolynes and his team at the university’s Center for Theoretical Biological Physics (CTBP) have applied his energy landscape theory to proteins that are hard to view because they live and work primarily inside cell membranes.
The method should increase the technique’s value to researchers who study proteins implicated in diseases and possibly in the creation of drugs to treat them, he said.
The study appeared this week in the Proceedings of the National Academy of Sciences. Lead author Bobby Kim, a graduate student, and co-author Nicholas Schafer, a postdoctoral research associate, are both members of Wolynes’ Rice lab.
Membrane proteins are critical to such functions as photosynthesis and vision, among many others. They can also serve as a cell’s gatekeepers by deciding what may pass through, and also as its gates by helping transport nourishment from the outside and waste from the inside. Because of these multiple roles, they constitute a large percentage of drug targets.
While their function is clear, information about how they fold lags far behind what is available for globular proteins, Wolynes said. “This is strange because membrane proteins are about 30 percent of the genome,” he said.
Wolynes and his colleagues use raw genomic information to predict how strands of amino acids will fold into functional proteins by following paths of least resistance (aka the principle of minimal frustration) dictated by the energy associated with each “bead” in the strand. The closer a protein gets to its functional “native” state, the more stable it becomes. Wolynes’ pioneering theory graphically represents this energy as a funnel.
The researchers test their computer models by comparing them to the structures of actual proteins acquired through X-ray crystallography. Plenty of structures are available for globular folded proteins, which float around the body to carry out tasks essential to life.
But until recent years, similar structures for transmembrane proteins have been hard to come by because of the difficulty of isolating them for imaging without destroying them. Recent advances use a detergent to wash most of the membrane away from a protein of interest, Wolynes said. “It leaves a fatty layer around the protein but nevertheless gives a sort of coating that allows the whole molecule to form a crystal lattice later on,” he said.
Wolynes was inspired to study membrane proteins when he noticed that two widely used cell biology textbooks were in complete disagreement about how they folded.
“One of them, after listing all the rules, said, ‘This is evidence that it’s kinetically controlled.’ The other said, ‘This is evidence that it’s equilibrium-controlled.’ They’re written in that way of introductory textbooks where anything they tell you about, they act as if it’s absolutely certain. And they were in direct opposition.
“I would say I’m still not certain, but I think our work points much more in the direction that folding is thermodynamically (equilibrium) controlled, at least once the protein is stuck in the membrane.”
Kim and Schafer modified a protein-folding algorithm used by the Wolynes lab called the Associative Memory, Water-Mediated, Structure and Energy Model (AWSEM) to account for outside influences unique to membrane proteins, including the translocon mechanism that inserts partially folded proteins into a membrane, and the membrane itself.
With the algorithm, they successfully determined that thermodynamic funnels still seem to hold the upper hand in folding proteins inside a membrane, as they do for globular proteins.
“We had a database of membrane protein structures from many different labs and we were able to learn the parameters that were transferable between them,” Kim said. “These parameters specify how strongly two residues (the “beads”) should interact and take into account the surrounding environment. That allowed us to make predictions from the raw sequences.”
The researchers expect to fine-tune the AWSEM-membrane algorithm as more structures become available. “I don’t think we’re done learning about membrane interactions,” Wolynes said, suggesting that much of the funneled folding happens after the protein enters the membrane and that very little of it is due to the hydrophobic (kinetic) interactions that play a somewhat larger role in globular protein folding. “My gut feeling is that’s going to be right,” he said.
“The significance of the paper is that we now have an algorithm to predict membrane protein structure pretty well based on the raw genome sequence,” Wolynes said. “This is going to be very useful to interpret a new generation of experiments.”
The National Institutes of Health, through the National Institute of General Medical Sciences, the National Science Foundation (NSF)-supported CTBP and the D.R. Bullard-Welch Chair at Rice University supported the research.
The researchers utilized the Data Analysis and Visualization Cyberinfrastructure (DAVinCI) supercomputer supported by the NSF and administered by Rice’s Ken Kennedy Institute for Information Technology.
Media Contact
All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
Red light therapy for repairing spinal cord injury passes milestone
Patients with spinal cord injury (SCI) could benefit from a future treatment to repair nerve connections using red and near-infrared light. The method, invented by scientists at the University of…

Insect research is revolutionized by technology
New technologies can revolutionise insect research and environmental monitoring. By using DNA, images, sounds and flight patterns analysed by AI, it’s possible to gain new insights into the world of…

X-ray satellite XMM-newton sees ‘space clover’ in a new light
Astronomers have discovered enormous circular radio features of unknown origin around some galaxies. Now, new observations of one dubbed the Cloverleaf suggest it was created by clashing groups of galaxies….

