First-ever toolkit for RNA sequencing analysis using a ‘pantranscriptome’
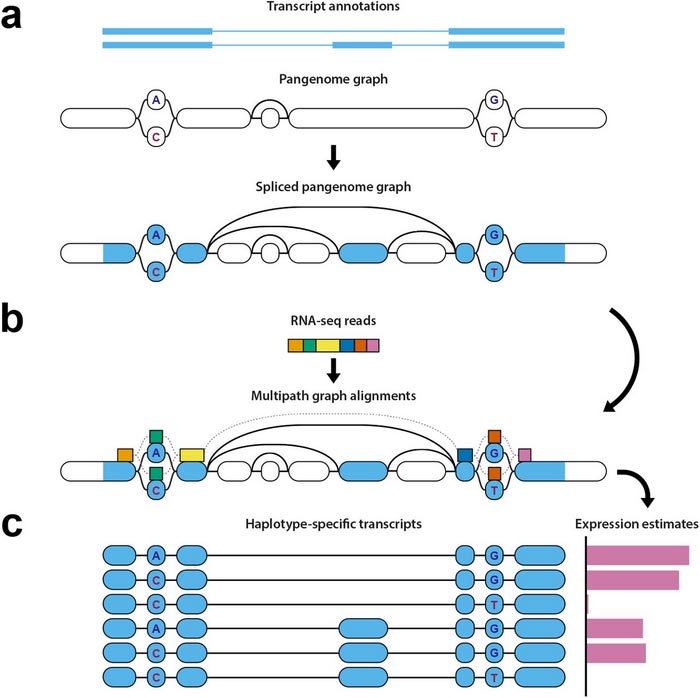
Diagram of haplotype-aware transcriptome analysis pipeline
Credit to study authors
Analyzing a person’s gene expression requires mapping their RNA landscape to a standard reference to gain insight into the degree to which genes are “turned on” and perform functions in the body. But researchers can run into issues when the reference does not provide enough information to allow for accurate mapping, an issue known as reference bias.
In a new paper published in the journal Nature Methods, researchers at UC Santa Cruz introduce the first-ever method for analyzing RNA sequencing data genome-wide using a “pantranscriptome,” which combines a transcriptome and a pangenome – a reference that contains genetic material from a cohort of diverse individuals, rather than just a single linear strand. A group of scientists led by UCSC Associate Professor of Biomolecular Engineering Benedict Paten have released a toolkit that allows researchers to map an individual’s RNA data to a much richer reference, addressing reference bias and leading to much more accurate mapping.
“This is pangenome plus transcriptome – that combination has never really been done before until now,” said Jordan Eizenga, the paper’s co-first author and a postdoctoral scholar in the UCSC Computational Genomics Lab. “This is the first time anyone has attempted to incorporate the pangenome as a standard feature of the RNA sequencing mapping.”
This tool will aid researchers around the world who are working to understand gene expression through RNA sequencing analysis. The tools are publicly available and can be accessed via Github.
“With this toolkit, we are employing this more diverse data that we can now get from the pangenome to improve the measurement of gene expression data, something that can widely vary between individuals,” Paten said. “The aim is to make the impact of this more diverse data felt on studies that are looking at gene expression, resulting in better analysis for cell models, organoid models, and other research applications.”
RNA’s most commonly recognized function is to translate DNA into proteins, but scientists now understand that the vast majority of RNA is noncoding and does not make proteins, but instead can play roles such as influencing cell structure or regulating genes. The entire RNA landscape is known collectively as the transcriptome, and mapping this allows researchers to better understand an individual’s gene expression.
The pantranscriptome builds on the emerging concept of “pangenomics” in the genomics field. Typically when evaluating an individual’s genomic data for variation, scientists compare the individual’s genome to that of a reference made up of a single linear strand of DNA bases. Using a pangenome allows researchers to compare an individual’s genome to that of a genetically diverse cohort of reference sequences all at once, sourced from individuals representing a diversity of biogeographic ancestry. This gives the scientists more points of comparison for which to better understand an individual’s genomic variation.
Mapping RNA sequencing data to understand gene expression can be difficult because the RNA sequences are spliced by cellular mechanisms, meaning one set of RNA data can come from non-connected areas of the genome, making it challenging to correctly align them to a reference. These splicing sites are not uniform across the human population, but vary between individuals. It is also difficult to know which haplotype the RNA comes from – whether the group of genes comes specifically from the set of chromosomes inherited from the individual’s mother, or the set inherited from the father.
But with the new pipeline of open source tools, the researchers can take the spliced segments of an individual’s RNA, map where they align on a pangenome, identify which haplotype the data belongs to, and analyze gene expression.
First, the pipeline identifies which areas of the genome the RNA sequencing data comes from, including the splice sites, and marks those points on the pangenome reference. Those marked points are then compared to a pantranscriptome consisting of haplotype-specific transcripts generated from the reference data contained within the pangenome. This step requires specialized, challenging algorithmic methods.
Finally, it generates estimates of levels of gene expression based on this comparison between the mapped data and the transcripts in the pantranscriptome, and identifies which haplotypes the genes come from.
“It’s definitely a very forward-looking study in that other genome-wide expression methods are not yet really utilizing pangenomes and haplotype information,” said Jonas Sibbesen, co-first author on the study and a former postdoctoral scholar in the UCSC Computational Genomics Lab who is now an assistant professor at the University of Copenhagen. “We’re now thinking ahead as to what pangenomics might additionally bring to the table in transcriptomic analyses.”
Going forward, the researchers are interested in further developing these tools to be useful for downstream informatics analysis, and tailoring the tools for the particularities of research on single-cell data. For now, the group hopes their new toolkit will serve to show how useful using pangenomics-derived analysis can be.
“We need to be able to explain to some researchers how a pangenome reference will benefit them,” Paten said. “This pipeline is really a first go at doing this for RNA, for functional data, for expression data.”
Journal: Nature Methods
DOI: 10.1038/s41592-022-01731-9
Article Title: Haplotype-aware pantranscriptome analyses using spliced pangenome graphs
Article Publication Date: 16-Jan-2023
All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.




Newest articles

“Nanostitches” enable lighter and tougher composite materials
In research that may lead to next-generation airplanes and spacecraft, MIT engineers used carbon nanotubes to prevent cracking in multilayered composites. To save on fuel and reduce aircraft emissions, engineers…

Trash to treasure
Researchers turn metal waste into catalyst for hydrogen. Scientists have found a way to transform metal waste into a highly efficient catalyst to make hydrogen from water, a discovery that…
Real-time detection of infectious disease viruses
… by searching for molecular fingerprinting. A research team consisting of Professor Kyoung-Duck Park and Taeyoung Moon and Huitae Joo, PhD candidates, from the Department of Physics at Pohang University…

