New gene for rare inherited paralysis may aid other spinal cord research, too
Discovery holds immediate promise for diagnosis & treatment of hereditary spastic paraplegia
A single mutation in a single gene is enough to slowly rob people of their ability to walk, scientists from the University of Michigan and the University of Pennsylvania report today.
And while the inherited defect itself is rare, its discovery may help researchers unravel the mysteries of much more common paralyzing conditions, from spinal cord injury to Lou Gehrig’s disease.
In a paper published online today in the American Journal of Human Genetics, and scheduled for the journal’s October issue, the U-M and Penn team describes a new gene, called NIPA1, for a form of hereditary spastic paraplegia, or HSP.
HSP is the name given to a group of disorders affecting about 20,000 Americans. HSP gradually disables its victims as long nerve cells in the spinal cord degenerate and muscles weaken and become spastic. It is often misdiagnosed as other nerve disorders, including multiple sclerosis, cerebral palsy, and amyotrophic lateral sclerosis (also called Lou Gehrig’s disease).
There is no cure for HSP, which is also sometimes called familial spastic paraparesis or Strumpell-Lorain disease. Treatment is limited to physical therapy and exercise to help retain as much muscle function as possible, drug treatment to tame spastic muscle movements, and medication to treat patients’ bladder and bowel control problems, and depression.
“There are direct overlaps between this group of diseases and the nerve degeneration processes in other disorders,” says senior author John Fink, M.D., professor of neurology at the University of Michigan Medical School, who has devoted his career to studying and treating HSP and related conditions. “Not only do we hope this discovery will aid HSP patients, but we also believe it will aid in understanding and perhaps stopping the fundamental processes involved in other types of spinal cord degeneration.”
The newly described gene mutation on chromosome 15 causes a form of HSP that starts in adulthood, and is passed on in families in a pattern called dominant inheritance. The paper’s co-lead authors are U-M research investigator Shirley Rainier, Ph.D., and Penn postdoctoral research fellow Jing-Hua Chai, M.D., who works in the laboratory of co-author Robert D. Nicholls, D.Phil.
The discovery of the mutation was made with the help of two families affected by the disease: one, of Iraqi origin living in Michigan, and the other of Irish descent, living in Arkansas. Affected and unaffected members of both families allowed their DNA to be studied and compared with DNA samples from people without HSP.
The single-nucleotide mutation alters the structure of a protein that the researchers think resides in nerve cell membranes, a feature that may expedite the development of tests and therapies. In addition to further research on the protein’s exact function, this gene discovery could be used to develop a genetic test to help give HSP patients and their families a firm diagnosis and genetic counseling. A provisional patent application has been filed jointly by the U-M and Penn.
The U-M team has previously found other genes involved in HSP, including a mutation responsible for a form of HSP that develops in early childhood and may resemble familial cerebral palsy.
That discovery, of a gene on chromosome 14 that encodes a protein called atlastin, has already yielded a diagnostic test that can give patients a firm diagnosis to tell them if they have or might some day develop that form of HSP. It can also help couples whose family or medical history suggests they might be at risk of passing a dominant form of HSP on to their children; in dominant HSP families, there is a 50 percent risk that each child will develop the condition.
Developed by Athena Diagnostics, which licensed U-M’s patent on the atlastin gene, the test also screens for a form of HSP found by French scientists, based on a gene for a protein called spastin. Fink has no direct financial interest in Athena Diagnostics.
Fink and Rainier have been looking at chromosome 15 for nearly eight years, after discovering that this region contained a mutant HSP gene. Rainier had narrowed the search to a very small region of chromosome 15, and carefully analyzed genes in this region as potential candidates. Meanwhile, Nicholls and his lab had been searching for genes in the same stretch of DNA.
Sharing of information and materials between the two laboratories resulted in the discovery that NIPA1 mutations cause this particularly severe form of HSP. In their paper, the researchers describe the gene and its encoded protein, as well as clinical analysis of affected subjects. The participation of 105 elderly men and women from a U-M Institute of Gerontology registry enabled this study, allowing the investigators to determine the range of normal variations of the new gene.
The affected members of the Iraqi family living in Michigan and the family from Arkansas are among the nearly 400 HSP patients that Fink and his U-M colleagues see each year. The U-M Neurogenetic Disorders Clinic is the largest clinical and research program for HSP and related disorders in the nation, and one of few that offer comprehensive evaluation, including genetic counseling.
The two families are not related, so the researchers believe that the same mutation occurred spontaneously in both, and likely causes the disease in other families and even in patients who have no family history of the disease.
Although more study is needed to understand exactly how the single-nucleotide mutation leads to a dysfunctional protein and thereby to the disabling symptoms of HSP, Fink and his colleagues feel that they have a good chance of unraveling the mystery. They’re working to develop a mouse model of this form of HSP, which will enable further studies of potential drug- and cell-based therapies.
Until this research yields new treatment options, the U-M and a few other centers offer treatment for HSP’s effects, and genetic testing and counseling. Support groups and a new Spastic Paraplegia Foundation bring patients and researchers together locally and nationally. Fink serves as the SPF’s medical advisor and last year hosted the first International Symposium for HSP.
In addition to Rainier, Chai, Fink and Nicholls, U-M laboratory assistant Debra Tokarz is a co-author. The research was supported by Fink’s Veterans Affairs Merit Review grant, the Geriatric Research, Education and Clinical Center of the VA Ann Arbor Healthcare System, and by Nicholls’ grants from the National Institutes of Health and the Muscular Dystrophy Association. American Journal of Human Genetics, Vol. 73, No. 4, Oct. 2003.
Media Contact
All latest news from the category: Health and Medicine
This subject area encompasses research and studies in the field of human medicine.
Among the wide-ranging list of topics covered here are anesthesiology, anatomy, surgery, human genetics, hygiene and environmental medicine, internal medicine, neurology, pharmacology, physiology, urology and dental medicine.



Newest articles
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
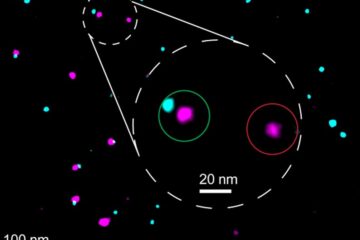
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

