Gene Mutations Responsible For Rett Syndrome In Females Present Sporadically in Males
In an international collaboration, researchers from the United States and Australia identified and evaluated four non-familial, sporadic occurrences of MECP2 gene mutations. Prior to this study, the majority of reported males with MECP2 mutations had a family history of RTT. Based on the results of this study, MECP2 abnormalities should be evaluated in young infant males who develop progressive encephalopathy including respiratory insufficiency, microcephaly, abnormal muscle tone and various movement disorders, as mutations to the gene may be the source of the infant’s neurological problems.
“Boys born to families with a history of Rett syndrome are examined very closely for MECP2 mutations, but beyond these families, physicians usually do not test for mutations to the gene,” said Walter E. Kaufmann, M.D., study author and research scientist at the Kennedy Krieger Institute in Baltimore. “Infant males with progressive encephalopathy may go undiagnosed because the prevailing assumption is that males with these mutations die before they are born. We’ve found that this is not the case, and encourage neonatologists and pediatricians to consider MECP2 as a possible cause of severe neurological abnormalities.”
All four newly identified cases exhibited common features, including: moderate or severe early postnatal progressive encephalopathy; unexplained central hypoventilation or respiratory insufficiency; abnormal movements; intractable seizures and abnormal tone. Three of the four cases had definitely pathogenic mutations and the fourth was potentially pathogenic. Acute observations and knowledge of the clinical picture of RTT prompted suspicion of MECP2 mutations in the four newly reported cases.
“While the findings of this study represent an important step forward in learning more about MECP2 mutations in infant males, many questions still remain regarding the role of the gene and its contribution to the encephalopathy of Rett syndrome,” said Dr. Gary Goldstein, President and CEO of the Kennedy Krieger Institute. “To help answer these and other questions regarding RTT, Kennedy Krieger has played a leading role in organizing an international consortium of scientists from every major Rett center around the world to conduct clinical studies on the diagnosis and treatment of the disorder.”
The consortium, called ‘RettSearch,’ will provide a forum for scientists to combine research efforts and share results from around the world. With only 15 cases of MECP2 mutations in infant males currently identified worldwide, researchers’ observations are limited. Through the ‘RettSearch’ network, which is being coordinated by Dr. Kaufmann, scientists from Kennedy Krieger and other member institutions hope to identify additional cases and conduct multi-center trials with both males and females with MECP2 mutations and RTT.
About Rett Syndrome
Rett Syndrome (RTT) is a neurological disorder often misdiagnosed as autism, cerebral palsy or non-specified developmental delay caused by a defective regulatory MECP2 gene found on the X chromosome. The disorder is seen almost exclusively in females. Unlike females, who have two X-chromosomes, males have an X and a Y chromosome. Because males lack a "backup" copy of the X chromosome that can compensate for a defective one, mutations in MECP2 are often lethal to the male fetus. This is why RTT is found overwhelmingly in females. RTT occurs in a variety of racial and ethnic groups worldwide and is now known to occur in 1:10,000 to 1:23,000 female births, but incidence may be far greater as new genetic evidence is discovered.
Development appears normal until 6-18 months of age, followed by loss of acquired speech and hand skills, slowing of head growth and development of stereotyped repetitive hand movements such as hand washing, hand wringing, hand tapping, hand clapping and hand mouthing. Stereotyped hand movements may change over time and additional problems may include seizures, breathing irregularities (hyperventilation and apnea), teeth grinding and curvature of the spine (scoliosis).
About the Kennedy Krieger Institute
Internationally recognized for improving the lives of children and adolescents with disorders and injuries of the brain and spinal cord, the Kennedy Krieger Institute in Baltimore, MD serves more than 12,000 individuals each year through inpatient and outpatient clinics, home and community services and school-based programs. Kennedy Krieger provides a wide range of services for children with developmental concerns mild to severe, and is home to a team of investigators who are contributing to the understanding of how disorders develop while pioneering new interventions and earlier diagnosis.
Media Contact
More Information:
http://www.kennedykrieger.org.All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles

Superradiant atoms could push the boundaries of how precisely time can be measured
Superradiant atoms can help us measure time more precisely than ever. In a new study, researchers from the University of Copenhagen present a new method for measuring the time interval,…
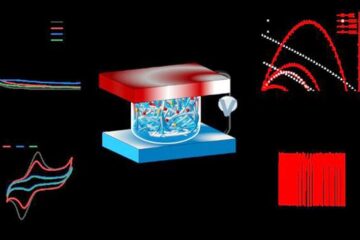
Ion thermoelectric conversion devices for near room temperature
The electrode sheet of the thermoelectric device consists of ionic hydrogel, which is sandwiched between the electrodes to form, and the Prussian blue on the electrode undergoes a redox reaction…

Zap Energy achieves 37-million-degree temperatures in a compact device
New publication reports record electron temperatures for a small-scale, sheared-flow-stabilized Z-pinch fusion device. In the nine decades since humans first produced fusion reactions, only a few fusion technologies have demonstrated…

