Biologist enhances use of bioinformatic tools and achieves precision in genetic annotation
In concrete, for his PhD thesis, “Strategies for the comparative genomic study of microorganisms with various levels of pregenomic information and genic complexity”, Dr Lavin used an emerging discipline: bioinformatics. By using informatics tools for studying sequences of DNA and proteins, he managed to generate databases for the genetic diversity of various organisms.
José Luis Lavín’s thesis has two differentiated parts which have in common the design of an enhanced technique of bioinformatic analysis that enables a greater degree of precision in the results. Thus, the first part of his work involved the study of bacteria, comparing genomes (the total set of genes in an organism) of pathogenic bacteria — the cause of illnesses —, both animal and vegetable. Through the comparison of genic sequences, gene candidates responsible for the infection process are detected. As the researcher himself explains, “We used very similar bacteria and looked for differences between them. In one study, we took a plant pathogen (Pseudomonas syringae) that attacks a number of crops such as bean, soya, tomato; in another we used various genomes of the causal agent for brucellosis (the Brucella genus), given that each variety of this bacteria attacks one type of animal breed specifically. In each case what was common was recorded and we have annotated what differentiated them”. These results are available to other research teams specialising in the treatment of bacteria so that they can use these target genes and, through mutations or simply by deactivating them, it can be seen if, effectively, they are responsible for plagues and illnesses and, thereby, control these.
During his stay in Denmark, in 2006, José Luis Lavín learnt the use of a new bioinformatic tool (HMM software), which he now uses in his research. The novelty, he explains, lies in that all the trials were carried out by computer. He used existing techniques but in such a way that he achieved the aim sought with the least possible percentage of error. The procedure has been considerably refined, given that, until now, the different techniques that existed were used separately and in an order that was not the correct one. Having defined parameters and a manual of correct procedure, very good results were achieved and with a minimum margin of error.
Sequencing of fungal genes
The second part of José Luis Lavín Trueba’s PhD focused on certain fungal genes, known as OXPHOS genes. These, if presenting defects in the human species, may produce illnesses such as Alzheimer, Parkinson’s, muscular dystrophies, etc. There are currently 27 genomes of fungi that have been sequenced and, amongst of all these what has been done is to detect what the differences are in the genes of the OXPHOS route. They did something similar as in the first part of the research with bacteria: they used bioinformatic tools and optimised them in order to achieve the detection of OXPHOS genes in the different genomes of the fungi; they looked for a methodology that enabled them to identify these genes with the greatest possible precision. In fact, this methodology is the reference pattern for a number of international projects for the sequencing of fungi genomes in which the Public University of Navarre research team also takes part. Fruit of this international collaboration is an article which has been accepted recently for publication in the PNAS (Proceedings of the National Academy of Sciences of the United States of America) journal, one of the most important publications in general science in the world today.
This unification of criteria and the procedure will enable greater precision for future researchers. The incorrect use of the tools that existed gave rise to anomalies – at times not detecting genes in a fungus when, in fact, they existed, or vice-versa. Dr Lavín explained with an example: “one of the bioinformatic tools, known as BLAST, makes a comparison of sequences: you can take a gene of a pine tree and send it to the database to see what it is similar to. Imagine that, of all the genes the database has identified, none are like that of the pine except a mouse gene, but in a minimal part. The tool would place this result in a prime position; but this did not mean the mouse gene is the same as the pine gene, simply that the two had a small part that was very similar, although 99.9% is very different. This is what happened until now: researchers assumed that, because a result came out first, it meant two genes were the same. In fact, a series of requisites has to be complied with and this is what the researcher has worked on in his thesis: identifying each of the genes that are truly similar or orthologous; i.e. they comply the same function or a very similar function in two different organisms.
Moreover, fruit of this research and, in collaboration with the Department of Mathematics at the Public University of Navarre, the development of the OXPHOS database is anticipated, and this will be available to the scientific community. What this means is that, when researchers need to carry out a study and search for these genes, they can refer to this database and, there being a high percentage of precision, they know the results are reliable. This database gathers the information of 27 fungi genomes already sequenced. The aim is that each time the genome of a new species of fungus is sequenced, the database incorporates this sequencing and the information available is extended.
Media Contact
All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
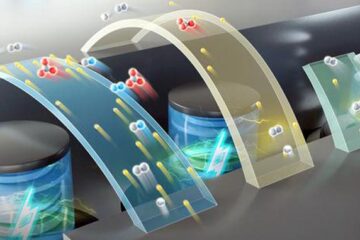
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
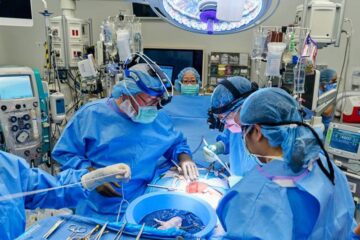
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

