Study reveals important new factor in cystic fibrosis lung inflammation
Cincinnati children’s study could lead to new therapeutics
Scientists at Cincinnati Children’s Hospital Medical Center have identified a missing piece of the puzzle of how lung damage occurs in cystic fibrosis (CF).
CF is a life-threatening, genetic disease that causes chronic lung infections and impairs digestion. The discovery, published in the current issue of Nature Immunology, provides impetus for the development of novel therapeutics that decrease inflammation in children with CF.
The Cincinnati Children’s team found a deficiency in the airways of children with CF of a class of molecules called lipoxins, which are key regulators of inflammation. “When we give analogs of this molecule (lipoxin-like molecules) in mouse models of CF, the molecules do what we’d like them to do — suppress acute inflammation, switch on the chronic inflammatory process and ameliorate disease — suggesting that this class of molecules might have therapeutic potential in CF,” says Christopher Karp, MD, director of Molecular Immunology at Cincinnati Children’s and the study’s main author.
Over time, the persistent combination of infection and inflammation in CF lungs leads to their destruction. Several studies in recent years suggest that the frequent and prolonged airway inflammation in CF lungs leads to the eventual cardio-respiratory failure that is the primary cause of death in people with CF.
“It’s traditionally been thought that the basic problem in the CF lung is an inability to clear bacteria, with infection leading secondarily to lung-damaging inflammation,” says Dr. Karp. “Recent studies suggest it may well be the other way around: abnormally vigorous and prolonged airway inflammatory may be a primary problem. Such responses are inefficient at clearing bacteria, may damage the airway in ways that promote colonization with bacteria, and over time lead to airway destruction.
“It’s clear that the inflammatory response of the airway in CF patients is abnormal from the earliest time you look at it. People haven’t traditionally thought of CF as an immunologic disease, but it is.”
Inflammation in the CF airway is acute inflammation — inflammation dominated by neutrophils, which are white blood cells that serve as a frontline protector against infectious pathogens. Normally, neutrophilic inflammation occurs for a short period of time. If the infection isn’t cleared, the inflammation shifts from being acute to chronic and involves different kinds of white blood cells, such as lymphocytes and macrophages.
The shift to chronic inflammation generally is a good thing, because chronic inflammation is less damaging to tissue. In patients with CF, however, the acute inflammatory response never changes to a chronic response. Lipoxins have been shown to play an important immunoregulatory role, acting as a key part of the switch that shifts the inflammatory response from acute to chronic.
“The more we know about the unique cycle of inflammation in CF, the better we are able to develop the appropriate tools to fight it. This study adds to our knowledge about CF and suggests an additional therapeutic direction,” said Robert J. Beall, Ph.D., president and CEO of the Cystic Fibrosis Foundation. “If we can reduce the rate at which lung tissue is damaged, we should be able to increase life expectancy and delay, or even eliminate the need for difficult and costly lung transplantation procedures.”
CF is a genetic disease affecting approximately 30,000 children and adults in the United States. A defective gene causes the body to produce an abnormally thick, sticky mucus that clogs the lungs and leads to life-threatening lung infections. These thick secretions also obstruct the pancreas, preventing digestive enzymes from reaching the intestines to help break down and absorb food.
People with CF have a variety of symptoms including: very salty-tasting skin; persistent coughing, at times with phlegm; wheezing or shortness of breath; an excessive appetite but poor weight gain; and greasy, bulky stools. Symptoms vary from person to person due, in part, to the more than 1,000 mutations of the CF gene. The median age of survival for a person with CF is in the early 30s.
Co-author of the study was Nicos Petasis, PhD, of the University of Southern California.
The study was funded by Cystic Fibrosis Foundation Therapeutics, the nonprofit drug discovery and development affiliate of the Cystic Fibrosis Foundation.
Cincinnati Children’s Hospital Medical Center is a 423-bed institution devoted to bringing the world the joy of healthier kids. Cincinnati Children’s is dedicated to transforming the way health care is delivered by providing care that is timely, efficient, effective, patient-centered, equitable and safe. It ranks third nationally among all pediatric centers in research grants from the National Institutes of Health. The Cincinnati Children’s vision is to be the leader in improving child health.
Media Contact
More Information:
http://www.cincinnatichildrens.org.All latest news from the category: Health and Medicine
This subject area encompasses research and studies in the field of human medicine.
Among the wide-ranging list of topics covered here are anesthesiology, anatomy, surgery, human genetics, hygiene and environmental medicine, internal medicine, neurology, pharmacology, physiology, urology and dental medicine.


Newest articles
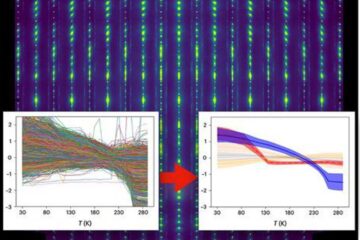
Machine learning algorithm reveals long-theorized glass phase in crystal
Scientists have found evidence of an elusive, glassy phase of matter that emerges when a crystal’s perfect internal pattern is disrupted. X-ray technology and machine learning converge to shed light…

Mapping plant functional diversity from space
HKU ecologists revolutionize ecosystem monitoring with novel field-satellite integration. An international team of researchers, led by Professor Jin WU from the School of Biological Sciences at The University of Hong…

Inverters with constant full load capability
…enable an increase in the performance of electric drives. Overheating components significantly limit the performance of drivetrains in electric vehicles. Inverters in particular are subject to a high thermal load,…

