It takes a sugar to catch a sugar
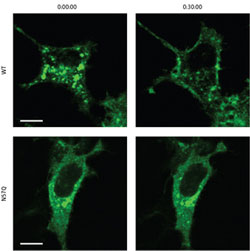
Figure 1: GLUT4 is normally retained within storage vesicles in the cytoplasm (top left), but is redistributed to the cell membrane within 30 minutes of insulin treatment (top right). However, the non-glycosylated N57Q mutant (bottom left) fails to respond to insulin and remains within cytoplasmic vesicles even after 30 minutes (bottom right) (scale bars, 10 ìm). <br>Copyright : 2011 American Society for Biochemistry and Molecular Biology <br>
After every meal, the hormone insulin is released into the bloodstream, issuing instructions to target cells to begin taking up excess sugar. In some situations, however, cells stop responding to these signals; and this insulin-resistant state is associated with onset of type 2 diabetes. Unexpected findings from Tadashi Suzuki’s group at the RIKEN Advanced Science Institute in Wako have now revealed how a cellular malfunction may contribute to this insulin resistance1.
Suzuki and postdoctoral fellow Yoshimi Haga had originally sought to develop imaging strategies to track localization of proteins modified with carbohydrate groups in a process known as glycosylation. They tested their method with the glucose-transporter protein GLUT4 and developed a mutant version of the protein that lacks a glycosylation site, but the results led their study in a new direction. “We accidentally found that behavior of our N57Q mutant and the wild-type protein was quite different,” says Suzuki. “It was a completely serendipitous finding.”
Insulin-responsive cells typically maintain reservoirs of GLUT4 in bubble-like vesicles within the cytoplasm; insulin signals induce the transport of GLUT 4 to the cell surface, where it begins pumping glucose into the cell. The N57Q mutant, on the other hand, was largely unresponsive to insulin (Fig. 1). Suzuki and colleagues determined that, instead of gathering within storage vesicles, this protein tended to accumulate either at the cell surface or within ‘recycling vesicles’ that continually shuttle to and away from the plasma membrane.
The N57Q mutant retains normal glucose-transporting capabilities, suggesting that this modification acts primarily as a trafficking signal rather than influencing protein function. Accordingly, the researchers observed the same insulin-insensitive behavior when they used a chemical treatment to alter the glycosylation of normal GLUT4. “These results clearly suggest that there must be a mechanism that detects subtle differences in glycan structure on this protein to sort it into specific GLUT4 vesicles,” says Suzuki.
Several studies have found evidence that GLUT4 may be subject to altered glycosylation in a subset of patients with type 2 diabetes; and, these new findings provide a potential explanation for how malfunctions in this protein modification process could contribute to pathology. As a next step, Suzuki hopes to uncover more details about how this transport pathway intersects with the insulin response. “We believe that glycan-recognition molecules known as lectins should be involved in this process,” he says, “and we will try to identify these lectins and other players involved in the fine-tuning of intracellular trafficking of GLUT4.”
The corresponding author for this highlight is based at the Glycometabolome Team, RIKEN Advanced Science Institute
Media Contact
All latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.




Newest articles

“Nanostitches” enable lighter and tougher composite materials
In research that may lead to next-generation airplanes and spacecraft, MIT engineers used carbon nanotubes to prevent cracking in multilayered composites. To save on fuel and reduce aircraft emissions, engineers…

Trash to treasure
Researchers turn metal waste into catalyst for hydrogen. Scientists have found a way to transform metal waste into a highly efficient catalyst to make hydrogen from water, a discovery that…
Real-time detection of infectious disease viruses
… by searching for molecular fingerprinting. A research team consisting of Professor Kyoung-Duck Park and Taeyoung Moon and Huitae Joo, PhD candidates, from the Department of Physics at Pohang University…

