RUB researchers unmask Janus-faced nature of mechanical forces with the Jülich supercomputer
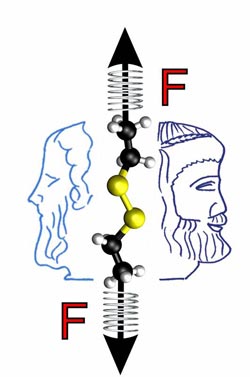
The Janus nature of mechanochemistry: Mechanical forces normally accelerate chemical reactions. However, in the case of disulfide bonds, which are present in large numbers in proteins, force-induced structural changes result in a relative deceleration above a certain threshold. The force thus shows its Janus-faced nature. Illustration: P. Dopieralski, D. Marx <br>
The harder you pull, the quicker it goes. At least, that used to be the rule in mechanochemistry, a method that researchers apply to set chemical reactions in motion by means of mechanical forces. However, as chemists led by Professor Dominik Marx, Chair of Theoretical Chemistry at the Ruhr-Universität Bochum now report in the journal “Nature Chemistry”, more force cannot in fact be translated one to one into a faster reaction.
With complex molecular dynamic simulations on the Jülich supercomputer “JUQUEEN” they unmasked the Janus-faced nature of mechanochemistry. Up to a certain force, the reaction rate increases in proportion to the force. If this threshold is exceeded, greater mechanical forces speed up the reaction to a much lesser extent.
Outdated view: mechanical force steadily reduces energy barrier
In order to activate chemical reactions, an energy barrier first has to be overcome. This energy can, for example, be supplied in the form of mechanical forces that “distort” the molecules involved. In order to achieve that experimentally, two long polymer chains are attached to the molecule. These chains serve as ropes to stretch the molecule either using a force microscope or by radiating the solution with ultrasound.
Until now it was assumed that the energy barrier decreases steadily, the more mechanical energy is put into the molecule. This hypothesis has now been refuted by the RUB-chemists. The key to success was a particularly complex form of computer simulation, the so-called ab initio molecular dynamics method, which they could only master on Europe’s currently fastest computer at the Jülich Supercomputing Centre within the framework of a “Gauss Large Scale” project.
Updated view: more force brings considerably less effect
The RUB team was looking at a small molecule with a disulfide bond, i.e. two sulphur atoms bound to each other, as a computational model in the “virtual laboratory”. “This molecule represents – in an extremely simplified fashion – the corresponding chemically reactive centre in proteins”, says Dominik Marx. In the course of the reaction, the sulphur bridge is cleaved.
The harder the chemists pull on the molecule, i.e. the more they distort the molecular structure, the faster the cleavage happens – but only up to a mechanical force of approximately 0.5 nanonewtons. Forces above ca. 0.5 nanonewtons accelerate the reaction significantly less than forces below this threshold.
Stressed molecules: too much mechanical force generates unfavourable spatial structure
The Bochum team could explain this effect based on the relative position of the individual molecular building blocks to each other. During the reaction, a negatively charged hydroxide ion (OH-) from the surrounding water attacks the sulphur bridge of the virtual protein. At forces above approximately 0.5 nanonewtons, however, the protein is already distorted to such an extent that the hydroxide ion can no longer reach the sulphur bridge without difficulties. The application of the force thus blocks the access, which increases the energy barrier for the reaction.
This can only be reduced again by an even greater mechanical force. In the next step, the researchers investigated the blockade mechanism on more complex models, including a large protein fragment, similar to previous experiments. “The Janus mechanism explains puzzling and controversial results of previous force-spectroscopy measurements on the protein titin, which is found in muscles”, says Prof. Marx.
Role of the solvent decisive for successful simulation
“Around the world, several theory groups have already tried to explain this experimentally observed phenomenon”, says Marx. “It was crucial to correctly take into account the role of the solvent, which is water in the present case.” The hydroxide ion that attacks the sulphur bridge is surrounded by a shell of water molecules, which changes over the course of the attack in a complex way.
The experimentally observed effects can only be correctly treated in the “virtual lab” when these so-called de- and re-solvation effects are accounted for included in the simulation as the reaction goes on. However, theorists usually resort to methods that drastically simplify the effects of the surrounding water (microsolvation and continuum solvation models) in order to reduce the computational cost.
Funding
The German Research Foundation (DFG) funded the study through what is so far the only “Reinhart Koselleck” project in the field of chemistry. In addition, the Cluster of Excellence “Ruhr Explores Solvation” (RESOLV, EXC 1069) has supported these studies since approval of the DFG in 2012. The project was only possible due to allocated computing time on the IBM Blue Gene/Q parallel computer JUQUEEN at the Jülich Supercomputing Centre. There, the Gauss Centre for Supercomputing (GCS) provided a large part of the total computation time within the framework of a “GCS Large Scale” project.
Bibliographic record
P. Dopieralski, J. Ribas-Arino, P. Anjukandi, M. Krupicka, J. Kiss, D. Marx (2013): The Janus-faced role of external forces in mechanochemical disulfide bond cleavage, Nature Chemistry, DOI: 10.1038/nchem.1676
Further information
Prof. Dr. Dominik Marx, Chair of Theoretical Chemistry, Faculty of Chemistry and Biochemistry at the Ruhr-Universität, 44780 Bochum, Germany, Tel. +49/234/32-28083, E-mail: dominik.marx@rub.de
Click for more
Chair of Theoretical Chemistry at the RUB
http://www.theochem.rub.de/home.en.html
Jülich Supercomputing Centre at the Research Centre Jülich
http://www.fz-juelich.de/ias/jsc/EN/Home/home_node.html;
jsessionid=F19FB1558F813DF7D80F04056353D9C2
Editorial journalist: Dr. Julia Weiler
Media Contact
More Information:
http://www.ruhr-uni-bochum.deAll latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.

Newest articles
Solving the riddle of the sphingolipids in coronary artery disease
Weill Cornell Medicine investigators have uncovered a way to unleash in blood vessels the protective effects of a type of fat-related molecule known as a sphingolipid, suggesting a promising new…

Rocks with the oldest evidence yet of Earth’s magnetic field
The 3.7 billion-year-old rocks may extend the magnetic field’s age by 200 million years. Geologists at MIT and Oxford University have uncovered ancient rocks in Greenland that bear the oldest…

Decisive breakthrough for battery production
Storing and utilising energy with innovative sulphur-based cathodes. HU research team develops foundations for sustainable battery technology Electric vehicles and portable electronic devices such as laptops and mobile phones are…

