New Technique Can Locate Genes’ On-Off Switches
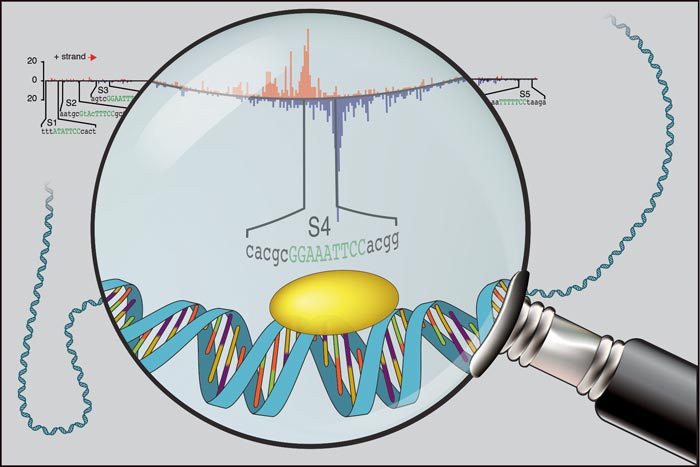
Image courtesy of the Zeitlinger Lab, Stowers Institute for Medical Research ChIP-nexus enables improved in vivo detection of transcription factors (yellow oval) on DNA, allowing to zoom in on individual binding footprints (data in red and blue).
In large part, this highly regulated process of gene expression is what makes us fully functioning, complex beings, rather than a blob of like-minded cells.
Despite its importance, researchers still do not completely understand how cells access the appropriate information in the DNA. They know this process is controlled by proteins called transcription factors, which bind to specific sites around a gene and – in the right combination – allow the gene’s sequence to be read.
However, functional transcription factor binding sites in the DNA are notoriously difficult to locate. The large number of transcription factors and cell types allow endless possible combinations, making it incredibly hard to determine where, when and how each binding event occurs. Moreover, results from genome-wide mapping efforts have only added to the confusion by suggesting that transcription factors bind very promiscuously all over the place, even to sites where they do not turn genes on or off.
Now, researchers at the Stowers Institute for Medical Research have developed a high-resolution method that can precisely and reliably map individual transcription factor binding sites in the genome, vastly outperforming standard techniques.
With the new technique, which was published March 9, 2015 in Nature Biotechnology, transcription factor binding sites that are likely functional leave behind clear footprints, indicating that transcription factors consistently land on very specific sequences. In contrast, questionable binding sites that were previously detected as bound showed a more scattered unspecific pattern that was no longer considered bound.
“Now we can see the subtleties, and a level of precision that we hadn’t anticipated,” says Stowers Associate Investigator Julia Zeitlinger, Ph.D., lead author of the study that also included Stowers colleagues Qiye He, Ph.D., and Jeff Johnston. “Not only do we see a distinct sequence motif where the transcription factor binds, but we also see additional sequences that seem to contribute to binding specificity. There is a lot more information that we can now read to understand how these factors act on the genetic code to influence expression.”
Over the last 15 years, a number of techniques have emerged to enable researchers to map where transcription factors bind to the genome. All of these techniques build on a method called chromatin immunoprecipitation or ChIP, which essentially tethers the proteins to their positions on the DNA, chops the DNA into manageable chunks, and then isolates the sections that are bound by the proteins.
Researchers have taken a variety of approaches to determine the sequence contained within these sections. ChIP-chip uses microarrays or gene chip technology to find the general neighborhood where a transcription factor’s footprint has appeared. ChIP-seq improves upon this approach by using the latest sequencing technologies, but still cannot pinpoint the exact address of the footprint.
The breakthrough came with ChIP-exo developed by Frank Pugh, Ph.D. and colleagues at Penn State University, which uses the addition of an enzyme called exonuclease to trim back the DNA fragments to the spot where the transcription factor is bound. Though this latest technique has promised to reveal the exact address of each transcription factor, its practical implementation had fallen short.
After several attempts to get the ChIP-exo technique to work in her laboratory, Zeitlinger decided to develop her own version. Having worked with ChIP-chip or ChIP-seq for over 15 years, Zeitlinger recognized that the much smaller amounts of DNA obtained by ChIP-exo made it very hard to obtain accurate sequence information. While helping a student working on an unrelated RNA technique, she saw a potential solution.
Normally, when researchers prepare a strip of DNA for analysis, they have to add an extra bit of sequence that serves as a kind of start site for the sequencing machinery. Traditionally, this prep involves two inefficient “ligation” steps, adding a bit of sequence first to the front and then to the back of each sample.
Zeitlinger and her colleagues figured out a way to accomplish the same feat in just one ligation step, adding a bit of sequence to the back of the fragment and then letting the strand form a circle. In addition, the researchers included a random bar code in the bit of DNA they used for ligation, which enabled them to catch any errors or artifacts that might arise in the sequencing procedure.
They called the new method “ChIP experiments with nucleotide resolution through exonuclease, unique barcode and single ligation” or ChIP-nexus. When they used ChIP-nexus to map the footprints of four well-known proteins – namely, human TBP and Drosophila NF-kappaB, Twist and Max— they found that it consistently outperformed existing ChIP-seq protocols in resolution and specificity.
The new tool could distinguish real footprints, those generated by a transcription factor sitting tightly on a particular sequence for a long time, from background noise, that may have arisen from a protein pausing on a sequence in its search for the right landing spot. Having a better collection of real footprints in turn provides much more detailed sequence information on the binding preferences of transcription factors.
Zeitlinger thinks the technique represents an important step forward for the field and will ultimately supplant ChIP-seq for the study of gene regulation.
“We still have a very simplistic idea of how transcription factors come in, open up the DNA, and turn on genes,” says Zeitlinger. ”If we do this kind of analysis for lots of transcription factors, we will gather information needed to better understand gene expression.”
In particular, she would like to see the technique used to ask how small changes in DNA — the kind that exist naturally in the human population — affect transcription factor binding and therefore differences in gene expression from one individual to the next.
The work was funded by the Stowers Institute for Medical Research and a grant from the National Institutes of Health (New Innovator Award 1DP2OD004561). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Lay Summary of Findings
At any given time, only a subset of the genes in a given cell are expressed or “turned on.” Proteins called transcription factors act as the molecular switchboard operators of the cell, binding specific sites in the DNA to flip different genes on and off. Despite their importance, researchers still have difficulty identifying these transcription factor binding sites. In the current issue of the scientific journal Nature Biotechnology, Stowers Institute scientists report the development of a new method called ChIP-nexus that can precisely and reliably map these sites, vastly outperforming previous techniques. Stowers Associate Investigator Julia Zeitlinger, Ph.D., who led the study, explains that researchers can use the new method to understand how transcription factors interact with DNA to control gene expression. For example, the patented technique has already shown that transcription factors binding sites are not scattered across the genome as previously thought, but rather appear in specific, predictable sequences.
About the Stowers Institute for Medical Research
The Stowers Institute for Medical Research is a non-profit, basic biomedical research organization dedicated to improving human health by studying the fundamental processes of life. Jim Stowers, founder of American Century Investments, and his wife, Virginia, opened the Institute in 2000. Since then, the Institute has spent over 900 million dollars in pursuit of its mission.
Currently, the Institute is home to almost 550 researchers and support personnel; over 20 independent research programs; and more than a dozen technology-development and core facilities.
Contact Information
Kim Bland
Head, Science Communications
ksb@stowers.org
816-926-4015
Media Contact
More Information:
http://www.stowers.orgAll latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.




Newest articles

Properties of new materials for microchips
… can now be measured well. Reseachers of Delft University of Technology demonstrated measuring performance properties of ultrathin silicon membranes. Making ever smaller and more powerful chips requires new ultrathin…

Floating solar’s potential
… to support sustainable development by addressing climate, water, and energy goals holistically. A new study published this week in Nature Energy raises the potential for floating solar photovoltaics (FPV)…
Skyrmions move at record speeds
… a step towards the computing of the future. An international research team led by scientists from the CNRS1 has discovered that the magnetic nanobubbles2 known as skyrmions can be…

