Several baffling puzzles in protein molecular structure solved with new method
The structures of many protein molecules remain unsolved even after experts apply an extensive array of approaches. An international collaboration has led to a new, high-performance method that rapidly determined the structure of protein molecules in several cases where previous methods had failed.
The usefulness of the new method is reported May 1 in Nature advanced online publication. The lead authors are Dr. Frank DiMaio of the University of Washington (UW) in Seattle and Dr. Thomas C. Terwilliger of Los Alamos National Laboratory in New Mexico. The senior author is Dr. David Baker, of the UW Department of Biochemistry.
A protein's molecular structure shapes its functions. In biomedical and health research, for example, scientists are interested in the molecular structure of specific proteins for many reasons, a few of which are:
To design drugs that selectively target, at the molecular level, particular biochemical reactions in the body
To understand abnormal human proteins in disease, such as those found in cancer and neurodegenerative disorders like Alzheimer's, and how these abnormal proteins cause malfunctions
To learn the shape and function of virus particles and how they act to cause infections
To see how the chains of amino acids, decoded from the DNA in genes, fold and twist into normally or abnormally shaped protein molecules
To design new proteins not found in the natural world, such as enzymes to speed up a slow biochemical reaction
To find ways to replace malfunctioning molecular parts of proteins that are critical to health
To devise nano-scale tools, such as molecular motors
“The important new method described this week in Nature highlights the value of computational modeling in helping scientists to determine the structures and functions of molecules that are difficult to study using current techniques,” said Dr. Peter Preusch, who oversees Baker's research grant and other structural biology grants at the National Institutes of Health (NIH). “Expanding the repertoire of known protein structures — a key goal of the NIH Protein Structure Initiative, which helped fund the research – will be of great benefit to scientists striving to design new therapeutic agents to treat disease.”
The methods devised by the group overcome some of the limitations of X-ray crystallography in determining the molecular structure of a protein. X-ray crystallography obtains information about the positions of atoms, chemical bonds, the density of electrons and other arrangements within a protein molecule.
The information is gleaned by striking protein crystals with X-ray beams. The beams bounce off in several directions.
Measuring the angles and intensities of these diffracted beams enables scientists to produce a 3-dimensional image of electron density. However, information about the molecular structure can be lost in taking the measurements, due to restraints posed by physics.
Scientists attempt to sidestep this problem by comparing the crystallography results to previously solved protein structures that resemble the unknown structure. The technique to “fill in the blanks” is called molecular replacement.
Molecular replacement has its own limitations in interpreting the electron density maps produced by X-ray crystallography, according to the authors of the paper. Techniques such as automatic chain tracing often follow the comparative model more closely than the actual structure of the protein under question. These mistakes lead to failure to obtain an accurate configuration of the molecule.
The researchers showed that this limitation can be substantially reduced by combining computer algorithms for protein structure modeling with those for determining structure via X-ray crystallography.
Several years ago, University of Washington researchers and their colleagues developed a structure prediction method called Rosetta. This program takes a chain of amino acids – protein-building blocks strung all in a row — and searches for the lowest energy conformation possible from folding, twisting and packing the chain into a three-dimensional (3-D) molecule.
The researchers found that even very poor electron density maps from molecular replacement solutions could be useful. These maps could guide Rosetta structural prediction searches that are based on energy optimization. By taking these energy-optimized predicted models, and looking for consistency with the electron density data contained in the X-ray crystallography, new maps are generated. The new maps are then subjected to automatic chain tracing to produce 3-D models of the protein molecular structure. The models are checked with a sophisticated monitoring technique to see if any are successful.
To test the performance of their new integrated method, the researchers looked at 13 sets of X-ray crystallography data on molecules whose structures could not be solved by expert crystallographers. These structures remained unsolved even after the application of an extensive array of other approaches. The new integrated method was able to yield high resolution structures for 8 of these 13 highly challenging models.
“The results show that structural prediction methods such as Rosetta can be even more powerful when combined with X-ray crystallography data,” the researchers noted. They added that the integrated approach probably outperforms others because it provides physical chemistry and protein structural information that can guide the massive sampling of candidate configurations. This information eliminates most conformations that are not physically possible.
Our procedures, the authors noted, required considerable computation, as up to several thousand Rosetta model predictions are generated for each structure. The researchers have developed automated procedures that potentially could narrow down the possibilities and lessen the number of times a model is rebuilt to make corrections. This automation could reduce computing time.
Through Baker's laboratory, many members of the general public contribute their unused home computer time to help in the effort to obtain structural models of proteins that are biologically and medically significant. The scientific discovery game is called “Fold It.” (http://fold.it/portal/)
Other authors of the paper appearing this week in Nature are Dr. Randy J. Read of the University of Cambridge, United Kingdom; Dr. Alexander Wlodawer of the National Cancer Institute; Drs. Gustav Oberdorfer and Ulrike Wagner of University of Graz, Austria; Dr. Eugene Valkov of the University of Cambridge; Drs. Assaf Alon and Deborah Fass of the Weizmann Institute of Science in Rehovot, Israel; Drs. Herbert L. Axelrod and Debanu Das of the SLAC National Accelerator Laboratory in Menlo Park, Calif.; Dr. Sergey M. Vorobiev of Columbia University in New York; and Dr. Hideo Iwai of the University of Helsinki, Finland.
The research was funded by the National Institute of General Medical Sciences and the National Center for Research Resources at the National Institutes of Health, the Howard Hughes Medical Institute, the Israel Science Foundation, DK Molecular Enzymology, Austrian Science Fund, the Center for Cancer Research at the National Cancer Institute, the Academy of Finland, and the U.S. Department of Energy's Office of Science, Biological and Environmental Research. The Joint Center for Structural Genomics, which is supported by the NIH's Protein Structure Initiative, contributed to the protein production and structural work.
Media Contact
More Information:
http://www.washington.eduAll latest news from the category: Life Sciences and Chemistry
Articles and reports from the Life Sciences and chemistry area deal with applied and basic research into modern biology, chemistry and human medicine.
Valuable information can be found on a range of life sciences fields including bacteriology, biochemistry, bionics, bioinformatics, biophysics, biotechnology, genetics, geobotany, human biology, marine biology, microbiology, molecular biology, cellular biology, zoology, bioinorganic chemistry, microchemistry and environmental chemistry.


Newest articles
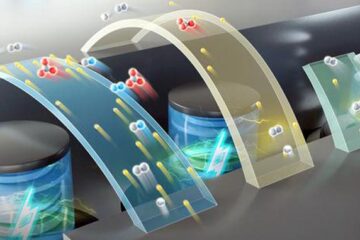
High-energy-density aqueous battery based on halogen multi-electron transfer
Traditional non-aqueous lithium-ion batteries have a high energy density, but their safety is compromised due to the flammable organic electrolytes they utilize. Aqueous batteries use water as the solvent for…
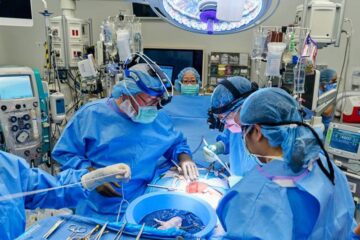
First-ever combined heart pump and pig kidney transplant
…gives new hope to patient with terminal illness. Surgeons at NYU Langone Health performed the first-ever combined mechanical heart pump and gene-edited pig kidney transplant surgery in a 54-year-old woman…
Biophysics: Testing how well biomarkers work
LMU researchers have developed a method to determine how reliably target proteins can be labeled using super-resolution fluorescence microscopy. Modern microscopy techniques make it possible to examine the inner workings…

